


Estructura y propiedades electrónicas de la nanoarcilla de caolinita dopada con metal de transición
Resumen
En este trabajo, se investigó una serie de nanoarcillas de caolinita dopadas con metales de transición (Cr, Mn, Fe y Co) mediante cálculos de la teoría funcional de la densidad (DFT). Se analizó la influencia del dopaje metálico en la estructura geométrica y la estructura electrónica de la caolinita. Se estudiaron los estados ferromagnéticos (FM), antiferromagnéticos (AFM) y no magnéticos (NM) de las estructuras de caolinita dopadas con metales de transición (TM). El volumen del cristal, los parámetros de la red, la longitud del enlace, la carga y el giro se calcularon mediante la teoría funcional de densidad corregida por dispersión (DFT-D2). Los resultados indicaron que Cr 3+ y Fe 3+ los dopantes se mostraron más estables en el estado AFM, mientras que Mn 3+ prefería los estados AFM y FM, y Co 3+ dopante prefería el estado NM. Además, el dopaje con metales de transición podría inducir la expansión del volumen de la red y algunos estados dopantes en la banda prohibida.
Antecedentes
Los minerales de nanoarcilla del grupo del caolín, como resultado de procesos de alteración hidrotermal y / o meteorización, tienen propiedades físicas únicas debido a su estructura en capas, tamaño de grano pequeño y, lo más importante, la superficie hidratada con muchos grupos hidroxilo. Ha atraído la atención de investigadores en química de materiales, química ambiental y física mineral [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11]. La caolinita, uno de los minerales de nanoarcilla más abundantes en la Tierra, se ha utilizado ampliamente en plásticos, catálisis y la industria del cemento. La mayor funcionalización de la caolinita como nuevos materiales de soporte ha atraído cada vez más atención en varios campos. La caolinita puede servir simplemente como material de soporte para mezclarse con otras nanopartículas para formar materiales de cambio de fase para la energía solar [4, 5] o recubierta con óxido dopado para formar polvos conductores para aplicaciones en campos conductores [9, 12]. Se descubrió que la hibridación de caolinita con nanopartículas funcionales mejora la actividad fotocatalítica de Pd-ZnO y las propiedades de luminiscencia de CdS a través de un efecto sinérgico [6, 7]. Las propiedades superficiales de la caolinita se modificaron anclando algunos grupos funcionales en la superficie [13, 14] o mediante un pretratamiento de activación ácida para una mejora adicional [2].
Las estructuras y la energía de los minerales del grupo del caolín se han investigado exhaustivamente de forma experimental [15,16,17] y teóricamente [18,19,20,21,22]. Se realizó un estudio teórico de la adsorción de metales pesados en la superficie de la caolinita para la adsorción de Cd, Cu, Hg y Ni (II) [23], en el cual la capacidad de adsorción de la arcilla caolinita para iones se encontró en el orden de Ni> Cu> Cd> Hg (II). Se estudió la adsorción y difusión de Pb (II) [24, 25] y uranilo [26] en la superficie de la caolinita (001) [24, 25, 26], y el comportamiento de adsorción en el sistema acuoso también se informó más tarde [27, 28]. La influencia del dopaje de Mg, Ca y Fe en la superficie de la caolinita y la posterior adsorción y penetración de H 2 Se estudiaron O en la capa intermedia [29]. Las energías de adsorción de H 2 O en caolinitas dopadas (001) se encontró menos que la superficie sin dopar. La estructura electrónica de la caolinita con y sin defectos intrínsecos ha sido estudiada por los funcionales y funcionales híbridos de la teoría funcional de densidad estándar (DFT) [30]. Sin embargo, hasta hace poco no se han modelado las evoluciones de la estructura durante el proceso de deshidroxilación, desaluminación y condensación de sílice de la caolinita mediante cálculos DFT [1, 31, 32]. La eliminación de Al en los materiales del grupo del caolín alteró en gran medida la geometría y las propiedades electrónicas de estos materiales de capa y mejoró su efecto de soporte [1, 2].
El dopaje de metales, como método bien conocido para modificar la estructura y las propiedades de los compuestos, se ha estudiado teóricamente para Al 2 O 3 [33], TiO 2 [34], MOF [35] y otros sólidos [36]. Sería interesante explorar los cambios en la estructura y las propiedades de la nanoarcilla de caolinita sobre el dopaje con metales de transición (TM) para este material de arcilla en capas. En este trabajo, se estudiaron una serie de nanoarcillas de caolinita dopadas con Cr, Mn, Fe y Co mediante cálculos DFT y se centraron en la influencia del dopaje metálico en la estructura geométrica y la estructura electrónica de la nanoarcilla de caolinita. Se estudiaron los posibles estados ferromagnéticos (FM), antiferromagnéticos (AFM) y no magnéticos (NM) de estas estructuras de caolinita dopadas con metales de transición. Los parámetros de la red, la longitud del enlace, la carga y el giro se optimizaron y calcularon mediante la teoría funcional de densidad corregida por dispersión (DFT-D2).
Métodos
Todos los cálculos se realizaron con el código del programa CASTEP (Cambridge Sequential Total Energy Package) [37], basado en el primer principio DFT. Para los cálculos se utilizó la aproximación de gradiente generalizada (GGA) con el potencial de correlación de intercambio de Perdew, Burke y Ernzerhof (PBE) [38]. Se incluyeron las correcciones de dispersión DFT-D2 de Grimme para tener en cuenta las interacciones de dispersión de Van der Waals [39]. Se aplicó un corte de energía de 500 eV utilizando el formalismo de onda plana de pseudopotencial ultrasuave [40]. La cuadrícula de Monkhorst – Pack [41] con 2 × 2 × 3 k Se utilizó una malla de puntos para la relajación geométrica y los cálculos de estructura electrónica. La energía total autoconsistente en el estado fundamental se obtuvo de manera efectiva mediante el esquema de mezcla de densidad [42]. Para las optimizaciones de geometría, el umbral de convergencia para la tolerancia de campo autoconsistente (SCF) se estableció en 1.0 × 10 −6 eV / átomo, todas las fuerzas sobre los átomos convergieron a menos de 0,03 eV / Å, el tensor de tensión total se redujo al orden de 0,05 GPa y el desplazamiento iónico máximo estaba dentro de 0,001 Å. Los elementos investigados en los estados de valencia fueron O (2s 2 2p 4 ), Al (3s 2 3p 1 ), Cr (3s 2 3p 6 3d 5 4s 1 ), Mn (3d 5 4s 2 ), Fe (3d 6 4s 2 ) y Co (3d 7 4s 2 ). Se utilizaron pseudopotenciales uspcc para Mn, Fe y Co, y pseudopotenciales usp para el resto de elementos. Los parámetros de la celda y la coordinación atómica se relajaron por completo durante la optimización de la geometría utilizando un algoritmo de minimización de Broyden-Fletcher-Goldfarb-Shanno (BFGS). La simetría del cristal se eliminó imponiendo diferentes momentos magnéticos iniciales en los iones TM para que el estado fundamental electrónico pudiera adoptar una simetría más baja.
Resultados y discusión
La estructura inicial de la caolinita se tomó para nuestro trabajo anterior [1]. La Figura 1 muestra la estructura cristalina relajada de 2 × 2 × 1 de la caolinita (4 unidades de caolinita). La estructura de la capa de caolinita, Al 2 Si 2 O 5 (OH) 4 , está compuesto por una hoja octaédrica de Al-O y una hoja tetraédrica de Si-O, conectadas por un átomo de O apical (O a ). El tetraedro Si – O está formado por un átomo de Si central y cuatro átomos de O circundantes, en el cual uno es el O a átomo y los otros tres son los átomos de O basales (O b ). El octaedro Al – O está construido por un Al central y seis O circundantes, en los que dos son O a átomo y los otros cuatro son átomos de O (en grupos OH) compartidos con otros octaedros de Al – O. Además, estos grupos OH se pueden dividir en dos tipos:la capa intermedia OH (OH inter ) en la superficie de la estructura de la capa y el OH interno (OH inner ) dentro de la estructura de capas entre la hoja de Al y la hoja de Si. Por lo tanto, hay dos tipos de enlaces Si – O, Si – O a y Si – O b (línea de puntos negra) y tres tipos de enlaces Al – O, Al – O inter (línea de puntos rojos), Al – O interior (línea de puntos verde) y Al – O (línea de puntos negros) en la estructura a granel de caolinita.
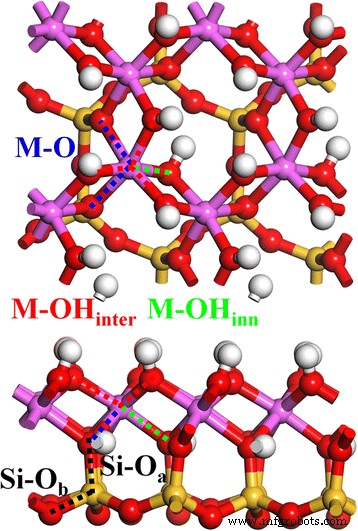
Arriba ( arriba ) y lateral ( hacia abajo ) vistas de caolinita. El Si – O a ( negro ), Si – O b ( negro ), M – OH inter ( rojo ), M – OH interno ( verde ) y M – O ( azul ) los enlaces se indican mediante líneas de puntos
La energía de dispersión siempre juega un papel importante en la estabilización de la estructura del mineral de arcilla debido a la interacción entre las capas [21, 43]. Entre los varios híbridos funcionales, PBE-D2 [21], B3LYP [22], B3LYP-D [18] y RPBE-D2 [18, 21], que se utilizó para obtener la estructura de celosía experimental de la caolinita [44, 45 ], Se encontró que el PBE-D2 funcional era preciso y requería menos tiempo. La sobreestimación del PBE funcional para las longitudes de los enlaces se supera mediante la corrección de la dispersión en comparación con los resultados experimentales, como se informó brevemente anteriormente [1]. Para distinguir el efecto del dopaje TM en la estructura de la caolinita, aquí revisamos primero la estructura de la red y las distancias de enlace optimizadas entre los cationes centrales (Si y Al) y los átomos de oxígeno, O a , O b y OH inn .
Como se muestra en la Tabla 1, para la caolinita, el volumen de celda unitaria calculado optimizado usando PBE-D2 funcional corregido por dispersión está cerca del valor experimental, lo que da un error relativo significativamente menor (∼0.4%) en comparación con PBE funcional (∼3.4%) . Para los vectores de celosía ayb, el error relativo usando PBE-D2 (∼0.4%) es mucho menor que PBE (∼1.1%). Y, con correcciones de dispersión de PBE-D2, la distancia de capa (vector c) de caolinita se reduce en 0,17 Å (∼2%). En particular, los ángulos de celosía después de la corrección de la dispersión están muy cerca de los resultados experimentales, especialmente para α. En cuanto a las distribuciones de longitud de enlace en caolinita, aunque PBE-D2 ofrece poca mejora para Si – O a , Al – OH interno , y los enlaces Al – O en comparación con los resultados experimentales, se logra una gran mejora para Al – OH inter unión en la superficie de Al – O (que es importante para la química de la superficie) y una ligera mejora para Si – O b unión en la superficie de Si – O. En particular, para Al – OH inter enlace, la corrección de dispersión de PBE-D2 parece describir con precisión el entorno de enlace en la capa más externa de la superficie de Al – O, que está fuertemente influenciada por la fuerza de dispersión de la superficie de Si – O de otra capa de caolinita que se encuentra arriba. Otro punto a mencionar aquí es que en realidad hay dos enlaces Al – O divididos (Fig. 1, línea de puntos azules) con longitudes de enlace significativamente diferentes de aproximadamente 1,95 y 2,00 Å [45], lo que muestra la distorsión reticular del Al – O El octaedro se originó a partir del desajuste de la red entre la hoja de Si-O y la hoja de Al-O. Como un error importante en el cálculo de la estructura de la caolinita en comparación con los resultados experimentales, estos enlaces Al – O están sobreestimados tanto por PBE como por PBE-D2, con una longitud de enlace promedio similar (Tabla 1). PBE-D2 da dos enlaces Al – O de aproximadamente 1,96 y 2,04 Å, con el segundo sobreestimado en 0,04 Å (Fig. 2, línea de puntos azul).
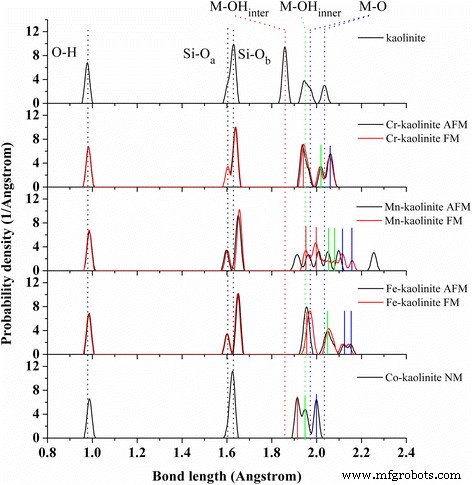
La distribución de enlaces de Cr–, Mn–, Fe– y Co – caolinita. Los estados multimagnéticos se dan para cada TM-caolinita. Los diferentes tipos promediados de O – H ( negro ), Si – O a ( negro ), Si – O b ( negro ), M – OH inter ( rojo ), M – OH interno ( verde ) y M – O ( azul ) los enlaces en la caolinita se indican mediante líneas de puntos . El M – OH inter ( rojo ), M – OH interno ( verde ) y M – O ( azul ) enlaces en Cr-caolinita ( AFM ), Mn – caolinita ( FM ), Fe-caolinita ( AFM ) y Cocaolinita ( NM ) se indican mediante líneas continuas
Las caolinitas dopadas con metales de transición (Cr, Mn, Fe y Co) se construyeron reemplazando el átomo de Al con un átomo de Cr, Mn, Fe o Co. Solo la sustitución equivalente de Al 3+ ion con TM 3+ Se consideró el ion ya que la sustitución no equivalente de los iones TM con un estado químico distinto de +3 provocará vacantes o impurezas adicionales para el equilibrio de carga. Desde el punto de vista de la estructura, las funciones PBE y PBE-D2 de TM-caolinita dan una diferencia de estructura similar a la observada para la caolinita. Considerando que PBE-D2 funcional describe mejor para vectores de celosía y longitudes de enlace de las dos superficies basales de caolinita, siguiendo la discusión sobre TM-caolinita, dependió principalmente de los resultados obtenidos por PBE-D2 funcional. Los parámetros de la red, la longitud del enlace, la carga y el espín de la caolinita dopada con TM y sus estados magnéticos se resumieron en la Tabla 1. Las diferencias de energía (por átomo de TM) entre los estados AFM y FM para Cr – caolinita, Mn – caolinita y Fe– caolinita son 0.022, -0.006 y 0.094 eV, respectivamente. Dado que la estructura de Co-caolinita solo es estable en un estado no magnético, solo se muestra la estructura NM de Co-caolinita.
Los volúmenes de celda unitaria de TM – caolinita se expanden en comparación con la caolinita, con una tendencia de Mn – caolinita> Fe – caolinita>> Cr – caolinita>> caolinita> Co – caolinita. Las expansiones celulares son causadas principalmente por los enlaces M – O más largos en comparación con los enlaces Al – O, lo que conduce a la mayor expansión en los vectores reticulares ay b. Mientras tanto, el Si – O b los enlaces en la hoja de Si – O se alargan simultáneamente, y los ángulos de la red cristalina de α y β se distorsionan en consecuencia. El volumen celular de Mn-caolinita con estado FM aumenta en un 1,4% en comparación con el estado AFM, mientras que, en contraste, se encuentra poca influencia del orden magnético en los volúmenes celulares para Cr-caolinita y Fe-caolinita. Los momentos magnéticos de Cr, Mn, Fe y Co son cercanos a los del Al 2 dopado con TM O 3 [33], mientras que la carga de Mulliken es ligeramente superior, lo que implica una reactividad más fuerte.
Las distribuciones de longitud de enlace de TM-caolinita se analizan en la Fig. 2, con diferentes tipos de enlaces Si – O y M – O en TM – caolinita indicados por líneas continuas para cada elemento de dopaje. Hablando en general, hay un aumento de las longitudes de enlace de M – O y Si – O b después del dopaje con TM, y mientras tanto hay una reorganización de la distribución de bonos de los enlaces M – O divididos para M – OH inter (rojo), M – OH interior (verde) y M – O (azul). En particular, los enlaces Al-O divididos (línea de puntos azules) desaparecieron después del dopaje con Cr y Co. Además, las distribuciones de la longitud de los enlaces dependen en gran medida del orden magnético de los átomos de Mn, pero solo están ligeramente influenciadas para los átomos de Cr y Fe.
Los resultados de PDOS para Cr 3+ (d3), Mn 3+ (d4), Fe 3+ (d5) y Co 3+ (d6) y las distribuciones de densidad de carga correspondientes se muestran en las Figs. 3 y 4. De acuerdo con el teorema de Jahn-Teller, cualquier sistema electrónico degenerado se distorsionará espontáneamente de tal manera que elimine la degeneración [46], que se ve afectada por el entorno de vinculación circundante [47]. Para TM 3+ dopaje en el sitio octaédrico de Al de la caolinita con muchos grupos hidroxilo, los cinco orbitales d-shell de TM 3+ se dividirá en un triplete t 2g estado y un doblete e g estado bajo Oh simetría. Los electrones en el estado triplete se localizan en la región media entre los ligandos y además se hibridan con los estados O más cercanos. Aquellos en el estado de doblete apuntan directamente a los ligandos y, por lo tanto, tienen una energía más alta que la t 2g electrones. Generalmente, la presencia de electrones en el e g Los orbitales tienden a desestabilizar el enlace octaédrico, y la degeneración se elimina alargando los enlaces opuestos al orbital lleno y acortando los enlaces opuestos al orbital vacío. La transición d – d de TM 3+ (Oh) la especie es siempre del t 2g ocupado orbitales (dxy, dyz y dzx) a e g desocupados orbital (d x2-y2 o d z2 , dependiendo de su ocupación). La división orbital entre e g orbitales y t 2g orbitales de Cr 3+ (d 3 ), Mn 3+ (d 4 ), Fe 3+ (d 5 ) y Co 3+ (d 6 ) en TM-caolinita es similar a la de Al 2 O 3 y TiO 2 [33, 48, 49], pero las energías de división entre los orbitales 3d son ligeramente mayores que en sus propios óxidos (Fig. 3), posiblemente debido a la hibridación con los grupos hidroxilo circundantes.
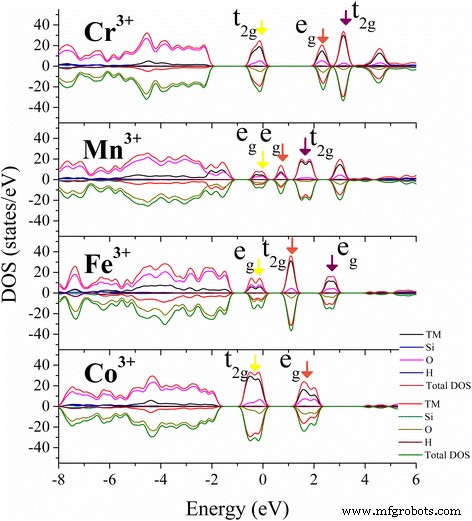
Se dan la densidad total de estados (DOS) y la densidad de estados proyectada por el átomo (PDOS) de los estados más estables para la caolinita dopada con TM. Los orbitales 3d ocupados más altos ( amarillo ) y el primero ( marrón ) y segundo ( violeta ) los orbitales 3d desocupados más bajos alrededor del nivel de Fermi están señalados por flechas de colores
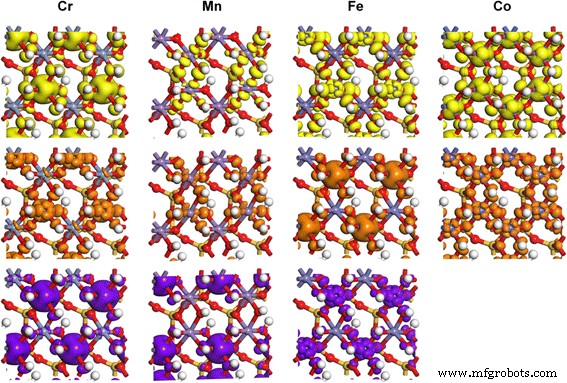
Densidad de carga parcial de los orbitales 3D de TM en TM-caolinita, correspondientes a los estados señalados por flechas en los resultados de PDOS. Los niveles de isosuperficie son 0.02 e / Å 3
La diferencia de energías de división entre los estados FM y AFM de Mn-caolinita es pequeña, y las distribuciones de densidad de estados son similares, excepto que las direcciones de giro son diferentes. Por lo tanto, por simplicidad, solo se muestran los resultados para el estado AFM. Para Mn 3+ de alto giro (d 4 ) ion en Mn-caolinita con estado AFM, solo uno de los dos e g los orbitales están ocupados en el máximo de la banda de valencia (VBM) (Fig. 3, flecha amarilla). La ocupación de d z2 El orbital que es más bajo en energía da una fuerte repulsión en los electrones de enlace de los dos ligandos a lo largo de la z eje y alarga el enlace M – O en esa dirección. Este efecto es el conocido efecto Jahn-Teller. Los estados en la parte inferior del mínimo de la banda de conducción (CBM) están compuestos por el menor desocupado d z2 orbital (flecha marrón) y el d x2y2 superior orbital (flecha violeta) de Mn 3+ (d 4 ). Para Cr 3+ (d 3 ), Fe 3+ (d 5 ) y Co 3+ (d 6 ) caso dopado, donde t 2g y e g los orbitales están ocupados uniformemente, la influencia del efecto de distorsión de Jahn-Teller es pequeña, lo que solo provocó una ligera desviación de los enlaces M-O en TM-caolinita (Fig. 2). Dicha modificación de la estructura y las propiedades electrónicas mediante el dopaje con TM podría mejorar la aplicación del caolín en el campo de la catálisis [50, 51], la captura de CO [52, 53], la carga de fármacos [54] y el almacenamiento de energía [55,56,57 ]. Y también se puede aplicar a otros minerales, como montmorillonita [50, 58], perlita [55] y talco [59] para alterar sus propiedades electrónicas.
Conclusiones
La influencia del dopaje con metales de transición (Cr, Mn, Fe y Co) sobre la estructura geométrica y la estructura electrónica de la nanoarcilla de caolinita se investiga mediante cálculos de DFT. Se calculan y estudian el volumen del cristal, los parámetros de la red, la longitud del enlace, la carga y el espín, y los posibles estados magnéticos. El Cr 3+ y Fe 3+ los dopantes se muestran más estables en el estado AFM, Mn 3+ prefiero el estado FM y Co 3+ los dopantes prefieren el estado de NM. El dopaje con metales de transición induce la expansión del volumen de la red y cierta reorganización de las distribuciones de los enlaces M – O. Mientras tanto, los dopantes TM introducen algunos estados 3d con energías de división más grandes en la banda prohibida de la caolinita.
Abreviaturas
- AFM:
-
Antiferromagnético
- BFGS:
-
Broyden – Fletcher – Goldfarb – Shanno
- CASTEP:
-
Paquete de energía total secuencial de Cambridge
- CBM:
-
Banda de conducción mínima
- DFT:
-
Teoría funcional de la densidad
- DFT-D2:
-
Teoría funcional de densidad corregida por dispersión
- FM:
-
Ferromagnético
- GGA:
-
Aproximación de gradiente generalizada
- NM:
-
No magnético
- PBE:
-
Perdew, Burke y Ernzerhof
- SCF:
-
Campo autoconsistente
- TM:
-
Metal de transición
- VBM:
-
Máximo de banda de valencia
Nanomateriales
- Presentación de la estructura atómica y electrónica de las nanofibras de carbono de copa apilada
- Modulación de las propiedades de anisotropía óptica y electrónica de ML-GaS por campo eléctrico vertical
- Estados electrónicos de nanocristales dopados con oxígeno y emisión visible en silicio negro Preparado por ns-Laser
- Influencia del agua en la estructura y propiedades dieléctricas de la microcristalina y nanocelulosa
- Propiedades ópticas y electrónicas de fotodiodos N + / P de silicio hiperdopado con azufre inducido por láser de femtosegundo
- Prueba de las propiedades estructurales, electrónicas y magnéticas de Ag n V (n =1–12) Clusters
- Morfología, estructura y propiedades ópticas de películas semiconductoras con nanoislinas GeSiSn y capas tensas
- Estructura electrónica y características I-V de las nanocintas InSe
- 20 tipos diferentes de metales y sus propiedades
- Cromo Metal:Elementos, Propiedades y Usos
- Propiedades de PCB automotriz y consideraciones de diseño