


Primer estudio de principios de penta-siligrafeno como material anódico de alto rendimiento para baterías de iones de litio
Resumen
A partir de los cálculos de los primeros principios, se predice que una nueva complejidad pentagonal de Si / C tendrá aplicaciones potenciales como material de ánodo prometedor para baterías de iones de litio. Se encuentra que la estabilidad estructural y térmica del penta-siligrafeno (P-Si 2 C 4 ) es mejor que el pentagrafeno, que se compone únicamente de átomos de C. El análisis de la estructura de la banda electrónica muestra que el C-2 p vacío z estado en el P-Si 2 C 4 proporciona espacio para acomodar y estabilizar electrones de Li, lo que hace que el almacenamiento de Li sea energéticamente favorable. Como resultado, cuatro átomos de Li pueden ser almacenados por una unidad de fórmula del P-Si 2 C 4 , correspondiente a una capacidad teórica de almacenamiento gravimétrico de Li de 1028,7 mAhg −1 . Las estructuras electrónicas metálicas del P-Li x adsorbido por Li Si 2 C 4 así como las barreras de energía de migración de Li muy pequeñas son beneficiosas para un rendimiento de carga / descarga rápida de la batería. El mecanismo de la interacción de adsorción de Li en el P-Si 2 C 4 se discute. Estos resultados demuestran una estrategia novedosa para diseñar materiales de ánodos complejos de Si / C bidimensionales para baterías de iones de litio de alto rendimiento.
Antecedentes
La densidad de energía relativamente baja de las baterías de iones de litio (LIB) comercializadas actualmente es difícil de cumplir con los requisitos de los vehículos eléctricos comerciales (EV) y se convierte en un gran desafío para el desarrollo de la industria de los vehículos eléctricos [1, 2]. Para mejorar la densidad de energía de los LIB, necesitamos mejorar la capacidad de los materiales de los electrodos. Debido a su muy buen rendimiento cíclico, el grafito es el material de ánodo más utilizado, pero su capacidad gravimétrica teórica (372 mAhg −1 ) es relativamente bajo [3, 4]. Por otro lado, el silicio tiene una capacidad gravimétrica teórica extremadamente alta de aproximadamente 4200 mAhg −1 [5], pero el rendimiento cíclico es pobre debido a su gran expansión de volumen hasta 420% en estado completamente litiado [6]. Para aprovechar las ventajas de los ánodos de silicio y carbono, diseñar un ánodo de complejo Si / C es tanto académicamente como tecnológicamente significativo.
El siligrafeno, que es un material en capas similar al grafeno bidimensional (2D) con átomos de C parcialmente reemplazados por átomos de Si, se predijo por primera vez que sería un material 2D estable a partir de los cálculos de los primeros principios [7,8,9,10] y se había preparado con éxito a partir de experimentos [11, 12]. Lin y col. habían demostrado que las láminas de SiC 2D se pueden preparar mediante técnicas de exfoliación en solución [11]. También prepararon con éxito SiC cuasi-2D 2 hojas que pueden conservarse en el aire durante meses [12]. Posteriormente, los cálculos de los primeros principios sugieren que el siligrafeno es un material de ánodo prometedor que ofrece una capacidad teórica de 1520 mAhg −1 y 1286 mAhg −1 para g-SiC 5 y g-SiC 2 , respectivamente [13]. Se muestra que el ánodo de siligrafeno hereda la alta estabilidad cíclica de los ánodos de grafito, así como la alta capacidad de los ánodos de silicio. La alta capacidad de almacenamiento de Li prevista se atribuye a la interacción mejorada de adsorción de Li con la monocapa de siligrafeno, que está relacionada con los cambios del átomo de Si de sp 2 a sp 3 -como [13]. Sin embargo, la configuración electrónica cambia de sp 2 a sp 3 -como se acompaña de cambios estructurales obvios durante la adsorción de Li en el siligrafeno. Esto no es bueno para el rendimiento cíclico del siligrafeno como material de ánodo para LIB. La mejor solución es diseñar materiales complejos de Si / C que ya tengan sp 3 -como configuración electrónica.
El carbono tiene muchos tipos de alótropos, que se forman con sp , sp 2 y sp 3 hibridación o sus combinaciones. El muy estable sp 2 Además, la configuración electrónica de enlaces π grandes en el carbono de grafito es responsable de las interacciones débiles de adsorción de Li en el grafeno. Tras la adsorción de Li en grafeno monocapa, se produce la transferencia de carga desde el Li a la capa de grafeno [14]. Luego, el Li se carga positivamente y se une a la capa de grafeno mediante la atractiva interacción de Coulomb. Sin embargo, el exceso de carga de Li en la capa de grafeno rompe el gran enlace π del grafeno, que es energéticamente desfavorable. Como resultado, la adsorción de Li en el grafeno de una sola capa no se ve favorecida con una energía de adsorción negativamente más alta que la energía cohesiva de la cara centrada en el cuerpo ( bcc ) fase de metal de litio, que no está permitido en baterías de iones de litio. Como resultado, no se permite el almacenamiento de Li con una monocapa de grafeno prístina [15]. Alternativamente, los materiales de carbono duro ofrecen una capacidad gravimétrica de almacenamiento de Li / Na mucho mayor en comparación con la de los materiales de carbono de grafito [16,17,18]. El material de carbono duro se conoce como una fase amorfa que se compone de sp 2 y sp 3 átomos de carbono [19]. ¿Es posible que la mayor capacidad gravimétrica de almacenamiento de Li de los materiales de carbono duro esté relacionada con la sp 3 configuración electrónica?
Pentagrafeno, que se conoce como sp 2 - sp 3 Se predijo que el carbono alótropo 2D híbrido [20] sería un material de ánodo prometedor para baterías de iones de litio / Na a partir de los cálculos de los primeros principios [21]. Como alótropo de carbono 2D, el pentagrafeno tiene un comportamiento de adsorción de Li mucho más fuerte en comparación con el grafeno convencional con estructura de panal. ¿Este comportamiento diferente de adsorción de Li también está relacionado con el sp 3 -como configuración electrónica en pentagrafeno? Si la respuesta es sí, ¿cuál es el mecanismo intrínseco detrás de esto?
Aunque se predice que el pentagrafeno es un alótropo de carbono dinámicamente estable, su energía cohesiva es significativamente mayor en comparación con la fase globalmente más estable (grafito o grafeno). La energía cohesiva del pentagrafeno es aproximadamente 0,9 eV por átomo más alta que la del grafeno hexagonal de una sola capa [20, 22], lo que dificulta (si es posible) la fabricación a gran escala del pentagrafeno industrialmente. Sin embargo, en cuanto a aplicaciones como material de ánodo, la fabricación a gran escala es muy importante. Tenga en cuenta que el pandeo se encuentra en el silicene y, por lo tanto, el Si es más estable con sp 3 -como hibridación que sp 2 [23,24,25] mientras que los átomos de C prefieren sp 2 hibridación en estructuras 2D; es razonable especular que reemplazar el sp 3 -al igual que los átomos de C con átomos de Si en la estructura del pentagrafeno se verán favorecidos energéticamente. A esta estructura la llamamos penta-siligrafeno. Experimentos recientes han demostrado que las nanocintas pentagonales basadas en Si se pueden cultivar en Ag (110) [26], lo que demuestra que la formación de una estructura pentagonal basada en Si es experimentalmente posible.
En teoría, la naturaleza electrónica y de enlace del penta-siligrafeno (P-SiC 2 ) fue estudiado por Lopez-Bezanilla et al. y encontraron que P-SiC 2 exhibe una inversión parcial del orden vertical de las bandas electrónicas p-p-σ y p-p-π [27]. Posteriormente, las propiedades de transporte electrónico del P-SiC 2 se estudian y comparan con pentagrafeno y penta-CN 2 [28]. Curiosamente, está demostrado que el rendimiento de transporte electrónico del P-SiC 2 se puede ajustar a través de la ingeniería de deformación, y se predijo que la deformación por compresión uniaxial es capaz de mejorar la movilidad del agujero de la monocapa penta-SiC 2 hasta 1,14 × 10 6 cm 2 V −1 s −1 [29]. A pesar de la similitud en las estructuras, el penta-siligrafeno tiene diferentes propiedades de transporte en comparación con el penta-grafeno. Fue encontrado por Hu et al. que la conductividad térmica del pentagrafeno exhibe una reducción monótona estándar por estiramiento, mientras que el penta-SiC 2 Posee un comportamiento inusual de subidas y bajadas no monótonas [30]. Estas interesantes propiedades del penta-siligrafeno están fuertemente relacionadas con la naturaleza electrónica y química de los átomos de Si en la estructura. También se encontró que el elemento Si en sí mismo es beneficioso para mejorar la adsorción de Li, ya que la interacción de adsorción de Li en el silicio es mucho más fuerte en comparación con la del grafeno [31, 32]. Por lo tanto, podría ser interesante saber si el penta-siligrafeno se puede utilizar como material de ánodo para LIB.
En este trabajo, investigamos los comportamientos de almacenamiento de iones de litio en penta-siligrafeno con cálculos de los primeros principios, y se analiza específicamente el mecanismo sobre cómo el penta-siligrafeno puede almacenar los iones de litio. Comenzamos nuestro estudio a partir de la estabilidad termodinámica del penta-siligrafeno, seguido de un análisis detallado de las interacciones intrínsecas de la adsorción de Li en él. Finalmente, se discute el desempeño del penta-siligrafeno como material de ánodo para LIBs.
Métodos computacionales
Todos los cálculos de este trabajo se realizan utilizando el Paquete de Simulación Ab initio de Viena (VASP) [33] basado en la teoría funcional de la densidad (DFT). Se utiliza el método de onda aumentada del proyector (PAW) [34, 35] combinado con el intercambio de aproximación de gradiente general (GGA) y las funciones de correlación parametrizadas por Perdew-Burke-Ernzerhof (PBE) [36]. La energía de corte para las ondas planas se elige en 450 eV para todos los cálculos. Los parámetros de la red y las posiciones iónicas están completamente relajados y las fuerzas finales convergen a 0.02 eV / Å. La estructura de la banda electrónica se calcula con el funcional híbrido Heyd-Scuseria-Erznerhof (HSE06) [37], ya que el funcional híbrido tiene una descripción más precisa de la estructura electrónica. El cálculo de la densidad de estados (DOS) se difumina mediante el método de difuminado gaussiano con un ancho de difuminado de 0,05 eV. El paquete Monkhorst [38] k se utiliza un muestreo de puntos y la densidad de k -malla es más gruesa que 0.05 Å −1 para simulación de dinámica de moléculas ab initio (AIMD) y 0,03 Å −1 para otros cálculos. La distribución de la carga atómica se analiza con el análisis de carga de Bader [39]. La ruta de migración de iones de litio se optimiza con el método de banda elástica con empuje de imagen ascendente (CINEB) [40]. La energía de adsorción E anuncio es calculado por:
$$ {E} _ {\ mathrm {ad}} =\ left ({E} _ {\ mathrm {host} + n \ mathrm {Li}} - {E} _ {\ mathrm {host}} - {nE } _ {\ mathrm {Li}} \ derecha) / n $$donde E host , E Li y E host + Li son las energías totales de los materiales de penta-siligrafeno del huésped, el átomo de Li y los huéspedes adsorbidos por Li, respectivamente, n denota el número de iones Li adsorbidos en el penta-siligrafeno. La influencia de las interacciones de van der Waals (vdW) en la energía de adsorción se prueba utilizando el método DFT-D3 con amortiguación de Becke-Jonson [41]. Además de la energía de adsorción, el potencial medio de intercalación de Li (frente a Li + / Li) se puede obtener directamente de la diferencia de la energía de adsorción y la energía cohesiva del metal Li (fase bcc) de V ave =- ( E anuncio - E Li - cohesivo ), si elegimos eV y V como unidades de energía y potencial, respectivamente.
Resultados y discusiones
Estructura y estabilidad del penta-siligrafeno
La estructura del pentagrafeno (ver Fig. 1a, denotado como P-C 6 a continuación de este documento) posee el P-42 1 m simetría (grupo espacial No. 113). Las constantes de celosía optimizadas son a = b =3.636 Å, de acuerdo con resultados anteriores [20, 21]. Se pueden encontrar dos tipos de átomos de carbono en la estructura, a saber, carbono con 4 coordenadas (indicado como C1 en la Fig. 1a) y carbono con 3 coordenadas (indicado como C2 en la Fig. 1a). De la geometría local de los átomos de carbono, podemos ver que C1 es sp 3 -como hibridado mientras que C2 es sp 2 -como hibridado. Aunque el átomo de C2 se considera sp 2 -como hibridado [20], la característica de enlace doble C2-C2 hace que el carácter químico del átomo de C2 sea diferente al del grafeno, que se discutirá en detalle a continuación de este artículo. Reemplazo de átomos de C1 con átomos de Si en el P-C 6 estructura, se forma el penta-siligraphene (ver Fig. 1b, el archivo de información cristalina de la estructura optimizada se da en el archivo adicional 1:SI-1 del material suplementario) y se indica como P-Si 2 C 4 a continuación de este documento. Como el radio atómico del átomo de Si es mayor que el del átomo de C, las constantes de red del P-Si 2 C 4 ( a = b =4.405 Å) es mayor que la de P-C 6 , mientras que está en buen acuerdo con los otros resultados reportados [27,28,29,30].
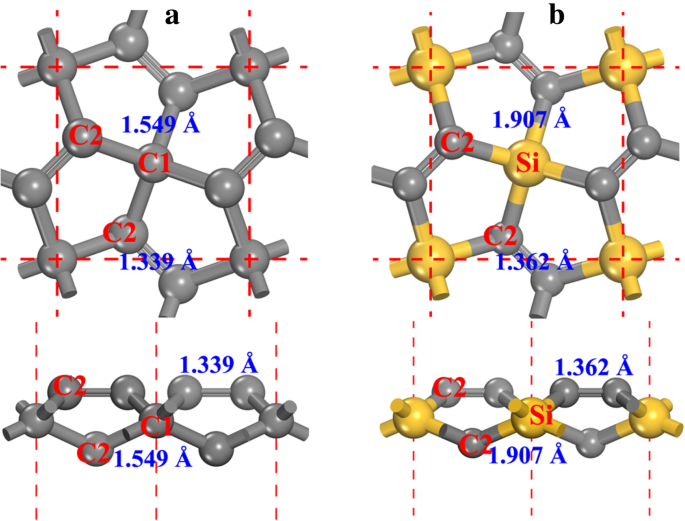
un El modelo de bola y palo de pentagrafeno y b penta-siligrafeno. Se presentan tanto las vistas superiores (arriba) como las laterales (abajo). Las esferas gris y amarilla son átomos de C y Si, respectivamente. Los átomos de carbono de 4 y 3 coordinados se denominan C1 y C2, respectivamente. Las longitudes de los enlaces también se presentan junto con cada enlace
Para evaluar la estabilidad termodinámica relativa, la Tabla 1 presenta las energías cohesivas de diferentes alótropos de complejos C, Si y C / Si. Aunque el pentagrafeno (P-C 6 ) se muestra estable a 1000 K a partir de simulaciones de dinámica molecular ab initio (AIMD) [20], la energía cohesiva de P-C 6 (- 8,24 eV · átomo −1 ) es mucho más alta en comparación con la del grafeno de una sola capa (- 9.14 eV · átomo −1 ). Esto muestra que la producción en masa de P-C 6 debe ser muy difícil. Por otro lado, la energía cohesiva de P-Si 2 C 4 (- 7,26 eV · átomo −1 ) es solo 0.2 eV más alto en comparación con su alótropo más estable g-Si 2 C 4 (- 7,46 eV · átomo −1 ), lo que demuestra que la preparación de penta-siligrafeno puede ser mucho más fácil en comparación con P-C 6 . Para verificar la estabilidad estructural del P-Si 2 C 4 , curvas de dispersión de fonones del P-Si 2 C 4 fueron calculados y presentados en la Fig. 2. Aunque pequeñas frecuencias imaginarias se encuentran en una pequeña región cerca del punto Γ (0.0039 THz o 0.13 cm −1 ), todavía podemos creer que el sistema es dinámicamente estable, porque generalmente se acepta que estas pequeñas frecuencias imaginarias (no mayores de 1 cm −1 ) podría ser un artefacto de la simulación [42]. También se han informado frecuencias imaginarias en otros materiales 2D dinámicamente estables como germanene [43] y arsenene [44]. Aplicando tratamientos técnicos como aumentar la precisión del cálculo o usar un método de cálculo diferente, estas frecuencias imaginarias se pueden eliminar.
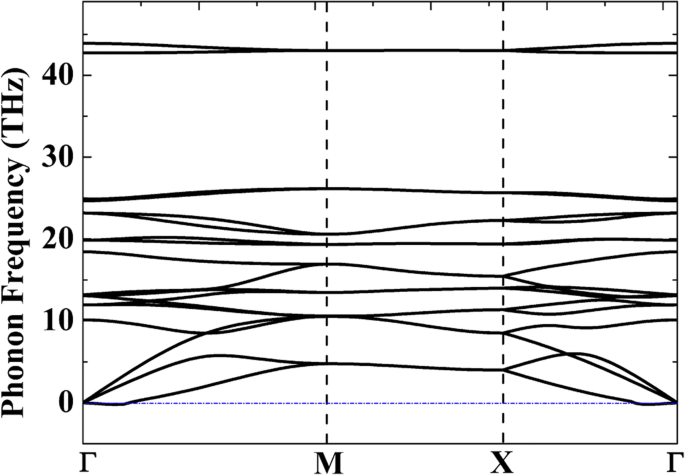
Curvas de dispersión de fonones del 2D P-Si 2 C 4 monocapa calculada a partir de la teoría de respuesta lineal
Además, la simulación AIMD también se realiza para evaluar la estabilidad estructural de P-Si 2 C 4 a altas temperaturas. Los AIMD se realizan utilizando supercélulas de 3 × 3 y 4 × 4 en un conjunto canónico a temperaturas de 1000 K, 1500 K, 2000 K y 2500 K (consulte el archivo adicional 1:Figura S1). Archivo adicional 1:Las figuras S2 y S3 presentan las configuraciones atómicas del P-Si 2 C 4 al final de las simulaciones AIMD a diferentes temperaturas utilizando supercélulas 3 × 3 y 4 × 4, respectivamente. Como se muestra, los anillos atómicos pentagonales se mantienen sin cambios cuando la temperatura es tan alta como 2000 K durante el tiempo de simulación de 20 ps, lo que muestra que la estructura puede soportar una temperatura tan alta como 2000 K. Por otro lado, deformaciones estructurales graves Se observan y anillos hexagonales (ver archivo adicional 1:Figura S2d), así como otros defectos (Archivo adicional 1:Figura S3d) aparecieron en las instantáneas, lo que indica que las estructuras están destruidas a 2500 K. Los anillos hexagonales encontrados en P-Si 2 C 4 a 2500 K indican que g-Si 2 C 4 (que se compone de anillos hexagonales [13]) es más estable que la penta-fase P-Si 2 C 4 , consistente con la energía cohesiva dada en la Tabla 1. Estos resultados confirman que la estabilidad estructural del P-Si 2 C 4 es mucho más estable en comparación con el P-C 6 , que solo puede soportar una temperatura de 1000 K.
Adsorción de Li en penta-siligrafeno
Para estudiar la adsorción de Li en el penta-siligraphene P-Si 2 C 4 , se consideran diferentes sitios de adsorción de Li y se pueden encontrar cuatro sitios de adsorción estables (como se muestra en la Fig. 3a) después de la relajación. Los sitios estables de adsorción de Li son el sitio superior del átomo de Si (indicado como T), el sitio hueco (indicado como H) del Si 2 C 3 anillo pentágono y sitios de puente entre dos átomos de C2 en la capa inferior (B1) y la capa superior (B2). La preferencia de la adsorción de iones de litio en estos sitios puede caracterizarse por las energías de adsorción presentadas en la Tabla 2. Los resultados muestran que el sitio de adsorción de Li más estable es el sitio B1, con una energía de adsorción de - 1.922 eV. Por otro lado, la energía de adsorción en el sitio H (-1,905 eV) está muy cerca del sitio B1. Las energías de adsorción de Li también están representadas por las alturas de adsorción, ya que una energía de adsorción más baja corresponde a alturas de adsorción más pequeñas (ver Tabla 2). Al comienzo del proceso de adsorción de Li, se prefiere que los iones de Li permanezcan en los sitios B1 más estables. Después de que todos los sitios B1 estén ocupados (correspondiente a una estequiometría de Li 2 Si 2 C 4 y ver Fig. 3b), los iones Li comienzan a permanecer en los sitios H. Como la distancia entre B1 y el sitio H es muy pequeña (~ 1,5 Å), se produce una fuerte interacción de repulsión de los iones Li en los sitios B1 y H. Como resultado, los iones de Li en los sitios B1 son rechazados hacia los sitios H cercanos y, por lo tanto, los sitios B1 quedan vacíos mientras que todos los sitios H están ocupados en el estado de Li 4 Si 2 C 4 (ver Fig. 3c). También se prueba la influencia de la interacción vdW en la energía de adsorción de Li, y los resultados se dan entre paréntesis en la Tabla 2. Como se muestra, la interacción vdW contribuye de - 0.12 a - 0.17 eV a la energía de adsorción para diferentes sitios de adsorción. mostrando que la interacción vdW está a favor de la adsorción de Li.
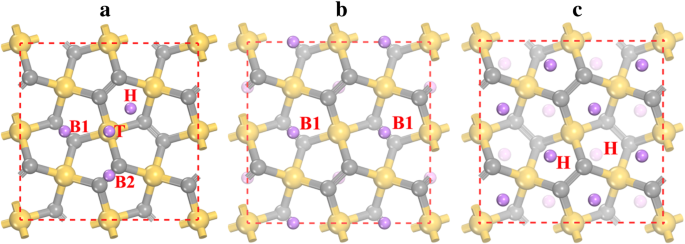
Sitios de adsorción de litio en a superficie de penta-siligrafeno y la configuración atómica de las estructuras más estables de b Li 2 Si 2 C 4 y c Li 4 Si 2 C 4 . Las esferas amarilla (más grande), gris (tamaño medio) y violeta (más pequeña) son átomos de Si, C y Li, respectivamente. H, T, B1 y B2 indican los sitios de adsorción de Li
Comparando las energías de adsorción de Li con la energía cohesiva del bcc fase Li metal (- 1,86 eV · átomo −1 ), podemos juzgar que la adsorción de Li es electroquímicamente activa o no. Si la energía de adsorción es menor que la energía cohesiva del metal Li, se favorece la adsorción de iones de litio y la adsorción corresponde a un potencial de descarga positivo. Como se indica en la Tabla 1, las energías de adsorción en los sitios B1 y H son inferiores a -1,86 eV, lo que muestra que los sitios B1 y H son sitios electroquímicamente activos para el almacenamiento de Li. Para evaluar la capacidad de almacenamiento de Li, las energías de adsorción de Li a diferentes concentraciones de iones de litio x (Relación Li / (Si + C)) se calculan y comparan con la energía cohesiva de bcc Li metal. Como se muestra en la Fig.4, las energías de adsorción de Li son inferiores a -1,86 eV cuando la relación Li / C x es menor que 2/3, correspondiente a 4 átomos de Li adsorbidos en una celda unitaria del P-Si 2 C 4 y una capacidad gravimétrica teórica de 1028,7 mAhg −1 (Li 4 Si 2 C 4 ). La densidad de energía de una batería, que es igual a la capacidad multiplicada por el voltaje de salida, está más preocupada en comparación con la capacidad de almacenamiento de Li. Un buen material de ánodo debe tener potenciales electroquímicos relativamente bajos, que pueden obtenerse de las energías de adsorción. El potencial promedio es de aproximadamente 0,1-0,2 V, que es relativamente bajo y beneficioso para un voltaje de salida más alto de un sistema de batería lleno. Además, la Fig. 4 también presenta los cambios de la constante de red a diferentes concentraciones de adsorción de Li. Como se muestra, la constante de celosía del P-Si 2 C 4 se encoge ligeramente con la adsorción de Li. Cuando la concentración x es 1/6, el cambio de la red alcanza el valor más grande, que es tan pequeño como - 0,94%, lo que indica que el cambio de volumen será muy pequeño durante el proceso de carga / descarga. El pequeño cambio de volumen es beneficioso para mantener la estructura del P-Si 2 C 4 para mantenerse estable durante el ciclismo.
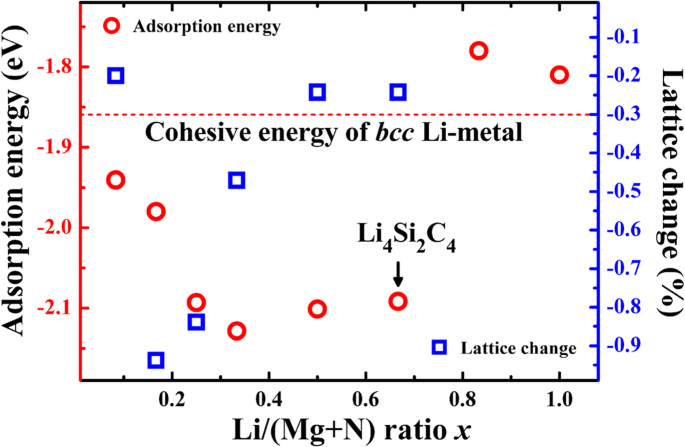
Las energías de adsorción de Li calculadas en P-Si 2 C 4 superficie (ciclos rojos, eje izquierdo) y el cambio constante de la red (cuadrados azules, eje derecho) en función de la concentración de adsorción de Li (relación Li / (C + Si)). La energía cohesiva de bcc El metal de fase Li también se incluye para comparación
Análisis de la estructura electrónica del penta-siligrafeno tras la adsorción de Li
Las estructuras de bandas electrónicas del penta-siligrafeno (P-Si 2 C 4 ) y sus estados litiados se presentan en la Fig. 5. Como se ve, P-Si 2 C 4 es un semiconductor con una banda prohibida indirecta de aproximadamente 2,35 eV, que es mucho más pequeña en comparación con los 3,46 eV del pentagrafeno P-C 6 (Ver archivo adicional 1:Figura S4). La banda prohibida más pequeña de P-Si 2 C 4 que P-C 6 se origina a partir de la dispersión mejorada de las bandas ocupadas más altas (números 11 y 12 en la Fig. 5a), particularmente en los puntos M y Γ de alta simetría. La banda prohibida se abre entre las bandas de energía del No. 12 (la banda ocupada más alta) y el No. 13 (la banda desocupada más baja). El nivel de energía de la banda No. 12 se eleva sustancialmente en el punto M, lo que eleva el nivel de Fermi y, a su vez, disminuye la banda prohibida.
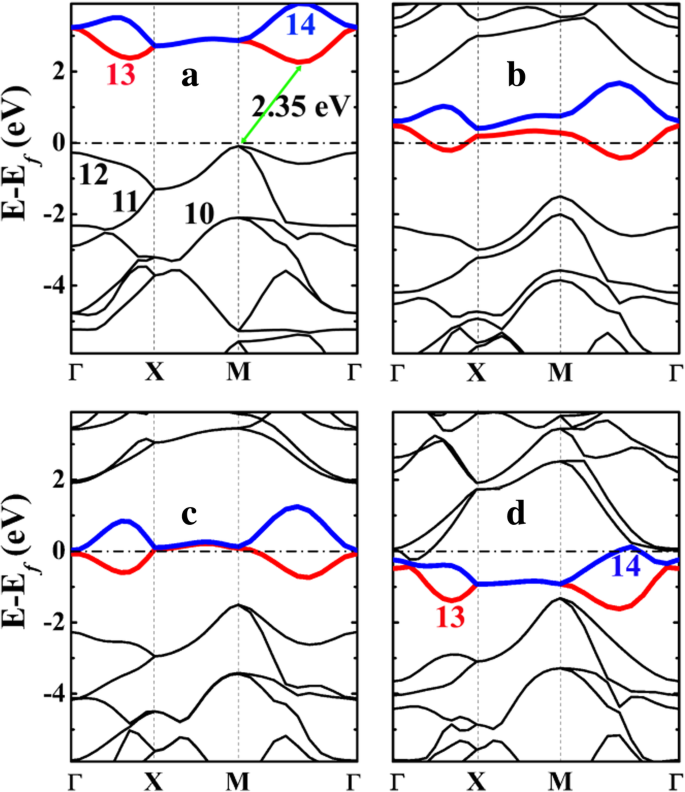
Las estructuras de bandas electrónicas de a penta-siligrafeno P-Si 2 C 4 y sus estados litiados b P-LiSi 2 C 4 , c P-Li 2 Si 2 C 4 y d P-Li 4 Si 2 C 4 calculado a partir de HSE06. El nivel de Fermi se selecciona para que sea 0 eV. Los números del 10 al 14 en a y d denota el número de la banda, mientras que los números de banda 13 y 14 están resaltados con colores rojo y azul, respectivamente. El etiquetado de las bandas se ajusta al código VASP, en el que el etiquetado de la banda se hace referencia a las bandas de valencia y conducción y los electrones del núcleo no se incluyen en el etiquetado
Del análisis de la forma de la densidad de carga (función de onda) proyectada a las bandas No. 10-14 que se muestran en la Fig. 6, podemos ver que la banda No. 12 corresponde a los estados de enlace de los enlaces σ formados entre C y Si átomos, mientras que la banda No. 13 (y 14) corresponde al 2- p z estado del átomo de C. La C- p vacía z El estado proporciona espacio para acomodar y estabilizar los electrones de la adsorción de Li, lo que hace que el proceso de adsorción de Li sea energéticamente favorable.
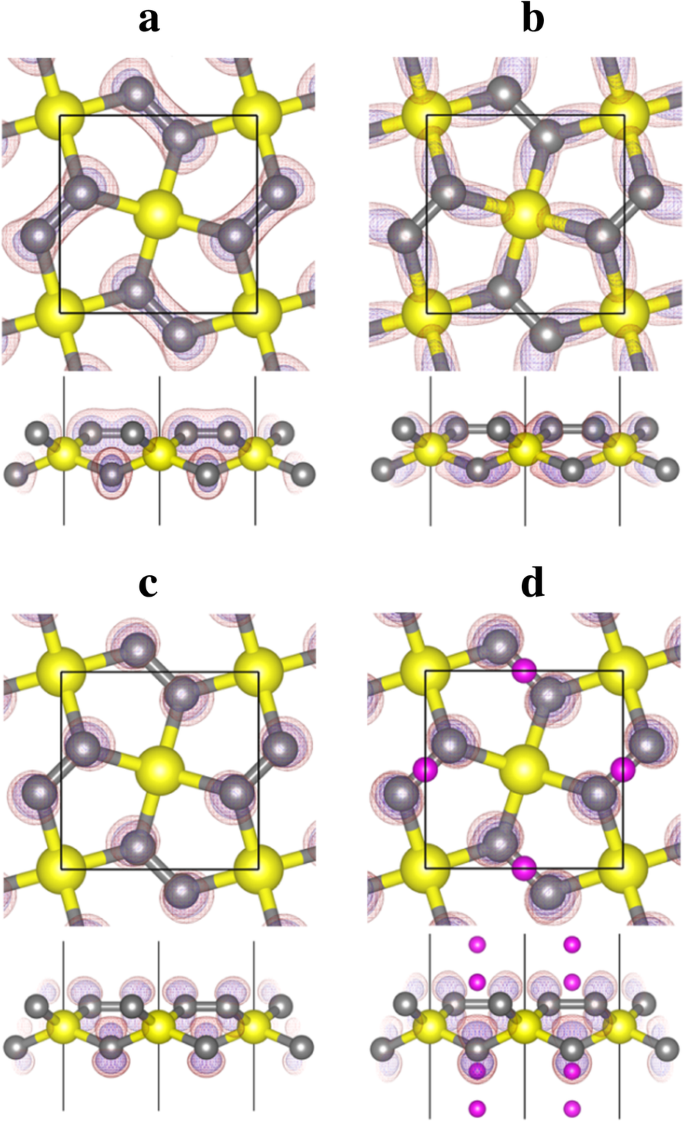
Contornos de densidad de carga descompuesta de banda para bandas a No. 10, b No. 12 y c No. 13 del penta-siligrafeno (P-Si 2 C 4 , Fig. 5a) y banda d No. 13 del penta-siligrafeno litiado (Li 4 Si 2 C 4 , Figura 5d). Las esferas amarilla (grande), gris (de tamaño medio) y violeta (pequeña) son átomos de Si, C y Li, respectivamente. Los contornos de densidad de carga se muestran en rojo transparente (con un valor de isosuperficie de 0,02 e / Å 3 ) y azul (con un valor de isosuperficie de 0,01 e / Å 3 ) colores
La dispersión mejorada de las bandas de energía ocupadas más altas en P-Si 2 C 4 puede atribuirse a dos factores:en primer lugar, la atracción de Coulomb entre los electrones que ocupan la banda n. ° 12 (enlaces C-Si σ) y el átomo de Si cargado positivamente, en comparación con la del pentagrafeno de carbono solo PC 6 . El análisis de carga de Bader muestra que el átomo de Si está cargado positivamente en el penta-siligraphene P-Si 2 C 4 . Como se muestra en la Tabla 3, la carga de Bader de átomos de Si en siligrafeno o penta-siligrafeno es de aproximadamente 1,65 e , que muestra que los átomos de Si están cargados positivamente con + 2,35 e . Por otro lado, el átomo de C1 en pentagrafeno (P-C 6 ) también tiene carga positiva, pero la carga neta es de solo + 0.08 e . Por lo tanto, además de la interacción de enlace covalente, se producen fuertes interacciones de Coulomb entre los átomos de C y Si en P-Si 2 C 4 , comparado con el del P-C 6 . Esto es beneficioso para la dispersión de las bandas de energía ocupadas (No. 12) cerca del nivel de Fermi. En segundo lugar, el pandeo mejorado en el P-Si 2 C 4 también puede contribuir a la dispersión de la banda No. 12 (el estado de unión del enlace σ C-Si), ya que un pandeo más grande denota más sp 3 -Se forman enlaces σ híbridos y más fuertes entre los átomos de C – Si. Esta es también una razón importante para la estabilidad estructural estable del P-Si 2 C 4 comparado con el de P-C 6 . Más importante aún, el pandeo entre los átomos de Si y C aumenta con la adsorción de Li y se convierte en 0.876 Å en el estado litiado P-Li 4 Si 2 C 4 . En este estado, los ángulos de enlace C – Si – C (102,50 ° y 124,59 °) del SiC 4 tetraedro se acerca a 108,47 ° de un tetraedro estándar, mostrando que el sp 3 -La hibridación del átomo de Si y la fuerza de los enlaces C-Si se vuelven más fuertes con la adsorción de Li. En consecuencia, la dispersión de la banda No. 12 también se mejora con el aumento de la concentración de adsorción de Li, como se puede ver en la Fig. 4.
Tras la adsorción de Li en la superficie del penta-siligrafeno (P-Si 2 C 4 ), transferencias de carga de los átomos de Li (en funcionamiento real con batería, Li + iones provienen del circuito interno mientras que la misma cantidad de electrones provienen del circuito externo) a los átomos de carbono en P-Si 2 C 4 . Como resultado, el exceso de electrones bajará por las bandas desocupadas (bandas No. 13 y 14) dando lugar a estructuras electrónicas metálicas del sistema, como se muestra en la Fig. 6b – d. Las estructuras electrónicas metálicas garantizan una buena conductividad electrónica del P-Si 2 C 4 ánodo durante el proceso de carga / descarga, que es beneficioso para el rendimiento de velocidad del sistema de batería utilizando el P-Si 2 C 4 ánodo.
Como se discutió anteriormente y se muestra por la banda de densidad de carga proyectada en la Fig.6, las bandas No. 13 y 14 son las p z estados del átomo de C en P-Si 2 C 4 . Estas bandas vacías son muy importantes para la adsorción de Li. Además de la atracción de Coulomb entre los iones Li cargados positivamente y el P-Si 2 cargado negativamente C 4 sustrato, los electrones que ocupan el C- p z los estados tienen una fuerte atracción de Coulomb hacia el átomo de Si cargado positivamente (que baja los niveles de energía de las bandas No. 13 y 14, y por lo tanto disminuye la energía total del sustrato). Como consecuencia, la adsorción de Li en P-Si 2 C 4 es energéticamente más favorable. Como celda unitaria del P-Si 2 C 4 contiene 4 átomos de C, se espera que 4 átomos de Li puedan ser adsorbidos en el P-Si 2 C 4 superficie. Después de la C- p z estados están completamente ocupados, adsorción de más átomos de Li en el P-Si 2 C 4 será energéticamente desfavorable. Esto está de acuerdo con las energías de adsorción calculadas que se presentan en la Fig. 4.
Dinámica de migración de iones de litio en P-Si 2 C 4
El rendimiento de velocidad del P-Si 2 C 4 El ánodo está determinado por la conducción electrónica y la dinámica de difusión de iones de litio. Como se discutió anteriormente, aunque la estructura electrónica del prístino P-Si 2 C 4 es un aislante, se vuelve metálico espontáneamente con la adsorción de Li, incluso cuando la concentración de Li es baja. Por lo tanto, la conductividad electrónica debe ser lo suficientemente buena para su aplicación como material de ánodo. Entonces, la difusión de iones de litio en el penta-siligrafeno se convierte en el paso de control de la velocidad. Como la estructura del P-Si 2 C 6 es similar al del pentagrafeno (P-C 6 ) en el que los iones de litio se difunden muy rápido [21], se espera que la difusión de iones de litio en P-Si 2 C 6 también puede ser muy rápido.
La tasa de rendimiento de una batería está fuertemente relacionada con el estado de carga (SOC), es decir, la difusión de iones de litio depende de la concentración de adsorción de Li. Para evaluar la dinámica de difusión de iones de litio en diferentes SOC, se consideran dos concentraciones extremas, a saber, iones de Li diluidos y vacantes de Li diluidos. Para la simulación de la difusión de iones de litio diluidos, se adsorbe un ión de litio en una supercélula del P-Si 2 C 4 . From above discussions in the section “Li Adsorption on Penta-siligraphene,” Li ion prefers to stay at B1 site when the Li concentration is low, as shown in Fig. 7. Considering the symmetry of the structure, only one Li migration pathway (denoted as Path-1 in Fig. 7) can be found and Path-1 forms a complete 2D Li diffusion network on the P-Si2 C4 superficie. The dilute Li-ion migration energy barrier on P-Si2 C4 along the NEB method optimized pathway is about 0.117 eV, which is smaller than that of on P-C6 (0.17 eV, Path-II) [21] and g-Si2 C4 (0.548 eV) [13]. When the SOC becomes 50%, namely, the material is discharged into the state of Li2 Si2 C4 , all Li ions still occupy the B1 sites (see Fig. 3b) and thus the Li-ion diffusion pathway is the same as the case of dilute Li ion. These results show that Li-ion diffusion can be very fast at the beginning half of the discharge process.

The dilute Li-ion migration pathway and the corresponding energy profile along the pathway on P-Si2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The red arrows illustrate the two-dimensional diffusion networks
For the case of dilute Li vacancy, we remove one Li ion from the fully lithiated state Li4 Si2 C4 and create one dilute Li vacancy in the supercell. As discussed above, Li ion prefers to occupy the H site when the Li concentration is high (see Fig. 3c). Therefore, three different Li vacancy migration pathways are considered, as shown in Fig. 8a. Path-1 refers to the Li vacancy migration from the H site to its neighboring H site across the top of a Si atom (across T site). Path-2 corresponds to the pathway across the top of the middle point of the C2–C2 dimer at the top layer (across B2 site). Path-3 is the pathway along the C2–C2 dimer at the down-layer (across B1 site). The energy profiles along the optimized pathways are given in Fig. 8b. As is seen, the Li-ion migration energy barriers along these pathways are very low, particularly for Path-3 (0.052 eV). The energy profiles along Path-1 and Path-2 are slightly asymmetric, because of the large relaxation of the Li ions when one Li vacancy is created. The extremely low energy barrier along Path-3 is reasonable, since Path-3 crosses B1 site (energetically most favorable adsorption site). However, Path-3 alone is not able to form a complete Li diffusion network on the surface of the P-Si2 C4 . Therefore, Path-1 or Path-2 must take part in the diffusion process, and the overall energy barrier for dilute Li vacancy migration is 0.155 eV or 0.165 eV. Although higher than dilute Li-ion migration (0.117 eV), the energy barrier for dilute Li vacancy migration is also very small compared with that for P-C6 (0.25 eV, Path-II’) [21] and g-Si2 C4 (0.233 eV) [13]. As the Li migration energy barriers in P-Si2 C4 are always lower than those in P-C6 and g-Si2 C4 (both dilute Li ion and dilute Li vacancy), it is expected that the rate performance of the P-Si2 C4 is the best one among the three similar anode candidates.

un Dilute Li vacancy migration pathways and b the corresponding energy profiles on fully lithiated P-Li4 Si2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The large green sphere represents the dilute Li vacancy. The thick/thin arrows indicate fast/slow migration pathways which form the two-dimensional diffusion networks
Conclusión
In summary, based on first principles calculations, we predicted that 2D pentagonal Si/C compound P-Si2 C4 can be potentially used as anode materials for LIBs. Phonon dispersion data confirmed the dynamic stability of the P-Si2 C4 structure at ground state, while AIMD simulation shows that the structure of the P-Si2 C4 can be stable at temperatures as high as 2000 K. The unique 2D buckled pentagonal structure promotes special empty C-2p z states that facilitate Li adsorption on the surface of the P-Si2 C4 , which offers a gravimetric Li storage capacity of 1028.7 mAhg −1 . The calculated dilute Li-ion/Li vacancy migration energy barriers show that Li-ion diffusion on the surface of the P-Si2 C4 can be faster than both the pentagonal graphene (P-C6 ) and the honeycomb-structured siligraphene. The metallic electronic structure of the lithiated P-Lix Si2 C4 ensures good electronic conductivity of the material as electrodes. These advantages are crucial features to the P-Si2 C4 as a promising anode material for LIBs. In summary, our first principles study offers a novel strategy to design high-performance Si/C complexity for the application in LIBs.
Disponibilidad de datos y materiales
All data generated or analyzed during this study are included in this published article.
Abreviaturas
- 2D:
-
Two-dimensional
- AIMD:
-
Ab initio molecule dynamics
- bcc :
-
Body-centered face
- CINEB:
-
Climbing image nudged elastic band
- DFT:
-
Density functional theory
- DOS:
-
Density of states
- EVs:
-
Electric vehicles
- GGA:
-
General gradient approximation
- HSE06:
-
Heyd-Scuseria-Erznerhof
- LIBs:
-
Li-ion batteries
- PAW:
-
Projector augmented wave
- PBE:
-
Perdew-Burke-Ernzerhof
- SOC:
-
State of charge
- VASP:
-
Vienna Ab initio Simulation Package
- vdW:
-
van der Waals
Nanomateriales
- Scalmalloy:el último material de alto rendimiento para impresión 3D en metal
- Nanocristales de estaño para futuras baterías
- Síntesis fácil de nanopartículas de SiO2 @ C ancladas en MWNT como materiales de ánodo de alto rendimiento para baterías de iones de litio
- Compuesto negro de acetileno / MoS2 de pocas capas como material de ánodo eficiente para baterías de iones de litio
- Preparación de micromateriales híbridos de MnO2 recubiertos de PPy y su rendimiento cíclico mejorado como ánodo para baterías de iones de litio
- Efecto de diferentes aglutinantes sobre el rendimiento electroquímico del ánodo de óxido metálico para baterías de iones de litio
- Na4Mn9O18 / Compuesto de nanotubos de carbono como material de alto rendimiento electroquímico para baterías acuosas de iones de sodio
- Compuesto de grafeno / Si integrado fabricado por reducción térmica de magnesio como material anódico para baterías de iones de litio
- Nanohojas V6O13 interconectadas en 3D cultivadas en textiles carbonizados a través de un proceso hidrotermal asistido por semillas como cátodos flexibles de alto rendimiento para baterías de iones …
- Un ánodo de película de Fe2O3 nanocristalino preparado por deposición de láser pulsado para baterías de iones de litio
- Síntesis e investigación de nanocables de CuGeO3 como materiales anódicos para baterías avanzadas de iones de sodio