


Conversación de MnBr2 antiferromagnético a monocapa ferromagnético de Mn3Br8 con MAE grande
Resumen
Una necesidad urgente en la espintrónica de baja energía son los ferroimanes bidimensionales (2D) con temperatura de Curie por encima de la temperatura del nitrógeno líquido (77 K) y una anisotropía magnética considerable. Estudiamos Mn 3 Br 8 monocapa que se obtiene mediante la inducción de vacantes de Mn en 1/4 de población en MnBr 2 monocapa. Esta configuración defectuosa está diseñada para cambiar la estructura de coordinación del Mn-d 5 y lograr ferromagnetismo con una considerable energía de anisotropía magnética (MAE). Nuestros cálculos muestran que Mn 3 Br 8 La monocapa es un semimetal ferromagnético (FM) con una temperatura de Curie de 130 K, un MAE grande de -2,33 meV por unidad de fórmula y un momento magnético atómico de 13 / 3μ B para el átomo de Mn . Además, Mn 3 Br 8 La monocapa se mantiene FM bajo una pequeña deformación biaxial, cuya temperatura de Curie bajo un 5% de deformación compresiva es de 160 K. Además, tanto la deformación biaxial como el dopaje del portador hacen que el MAE aumente, que principalmente contribuye por la energía de anisotropía magnetocristalina (MCE). Nuestra estructura defectuosa diseñada de MnBr 2 La monocapa proporciona una manera simple pero efectiva de lograr ferromagnetismo con MAE grande en materiales 2D.
Introducción
La espintrónica, que explota el espín del electrón y el momento magnético asociado, ha atraído una gran atención durante las últimas décadas [1], debido a sus ventajas únicas sobre los dispositivos basados en carga. La reciente realización de ferroimanes bidimensionales (2D) con ordenamiento magnético de largo alcance a temperatura finita [2, 3] es de gran importancia para la espintrónica a nanoescala y aplicaciones relacionadas e inspira enormes esfuerzos en las investigaciones y fabricaciones de ferroimanes 2D [4,5 , 6,7,8,9].
Los dos primeros ferroimanes 2D con espesor atómico se lograron en 2017, es decir, monocapa CrI 3 [2] y bicapa Cr 2 Ge 2 Te 6 [3]. Desafortunadamente, ambas temperaturas de Curie son más bajas que la temperatura del nitrógeno líquido (77 K), lo que limita sus aplicaciones realistas. Además de la temperatura de Curie, la anisotropía magnética considerable y el momento magnético también son indispensables para la aplicación práctica. La gran energía de anisotropía magnética (MAE) implica el beneficio del ordenamiento magnético contra la fluctuación del calor y la posibilidad de reducir el tamaño de grano por bit de información; pequeño MAE puede resultar en super-paramagnético en lugar de ferromagnético. El momento magnético grande proporciona una mayor sensibilidad, mayor eficiencia y mayor densidad para la espintrónica. Es más probable que los elementos pesados traigan un MAE grande debido a su fuerte efecto de acoplamiento espín-orbital (SOC) [10]. Se ha predicho que una serie de materiales 2D FM compuestos de elementos pesados tienen un MAE grande, como CrI 3 [11], CrAs [12], CrSeI [13], CrSiTe 3 [14], CrWI 6 [15], FeBr 2 y FeI 2 monocapas [16]. Además, el momento magnético local en el átomo de Mn de MXenes Mn 2 NF 2 y Mn 2 N (OH) 2 es 4.5μ B por átomo de Mn [17], que es el más grande entre los materiales FM 2D reportados.
Desde CrI 3 monocapa se ha sintetizado con éxito, los haluros de metales de transición han atraído mucha atención [18,19,20,21,22,23,24,25,26,27]. Se ha observado el efecto Spin Seeback en bicapa MnF 2 [20]; pocas capas de CrI 3 se ha implementado en las uniones de tunelización magnética (MTJ) [21]; NiCl 3 Se ha predicho que la monocapa será un nuevo semiconductor sin separación de espín (SGS) de Dirac [22]. En particular, MnBr 2 la monocapa es antiferromagnética con MAE de 0,25 meV a lo largo de la dirección perpendicular al plano según los cálculos de los primeros principios [16]; Mn 2+ los iones están en el d 5 estado de alto giro con momento magnético de 5μ B [16, 26]. Estos resultados implican los potenciales de MnBr 2 como ferromagnet monocapa con gran momento magnético. El problema clave es cómo convertir el acoplamiento AFM entre iones Mn en acoplamiento FM.
Se observó experimentalmente una densidad significativa de vacantes de Mn en LaMnO 3 películas delgadas [28], y la concentración de defectos se puede controlar regulando deliberadamente el proceso de síntesis mediante la irradiación de partículas de alta energía o el grabado químico [29]. En este contexto, diseñamos el Mn 3 Br 8 monocapa al inducir una sola vacante de Mn a MnBr 2 monocapa. La vacante cambia la estructura de coordinación del átomo de Mn y rompe el d 5 configuración, que puede convertir el acoplamiento antiferromagnético en acoplamiento ferromagnético y traer un MAE grande debido al átomo de Br pesado. Como esperamos, Mn 3 Br 8 La monocapa es FM y tiene un MAE grande de -2,33 meV por unidad de fórmula, el momento magnético para cada átomo de Mn es 13 / 3μ B . Considerando la fácil introducción de deformación mediante la flexión de sustratos flexibles [30,31,32,33], alargando el sustrato elástico [33,34,35], aprovechando el desajuste de expansión térmica [33, 36], etc. [33], y el control efectivo de la polarización de espín a través del dopaje electrostático [37, 38], también estudiamos el Mn 3 Br 8 monocapa bajo tensión biaxial y dopaje con portador. Nuestros resultados muestran que Mn 3 Br 8 La monocapa se mantiene FM con la temperatura de Curie aumentando bajo una pequeña tensión biaxial. Además, tanto la cepa biaxial como el dopaje con portador pueden hacer que el MAE aumente.
Métodos computacionales
Todos los cálculos del presente estudio se realizaron mediante la adopción del método de la teoría de la función de densidad con polarización de espín (DFT) implementado en el ab-initio de Viena. paquete de simulación (VASP) [39]. Las interacciones entre electrones y núcleos se describieron mediante el método de onda aumentada del proyector (PAW) [40, 41], y las interacciones de intercambio electrónico-correlación se describieron mediante el funcional Perdew-Burke-Ernzerhof (PBE) dentro de la aproximación de gradiente generalizado (GGA) método [42]. Se adoptaron los términos U de Hubbard para calcular la interacción fuertemente correlacionada [43]; Para los electrones Mn-d se utilizaron un parámetro de interacción coulomb eficaz en el sitio (U) de 4 eV y una energía de intercambio (J) de 1 eV que se adoptó para estudiar materiales 2D incorporados con Mn [44]. La integración de la zona de Brillouin se llevó a cabo adoptando la malla 9 × 9 × 1 k basada en el esquema Monkhorst-Pack [45]. Los espectros de fonones se calcularon utilizando el código Phonopy [46] que se implementa dentro del paquete VASP. Se añadió un espacio de vacío de 20 Å a lo largo de la dirección perpendicular a la superficie de la monocapa para evitar la interacción entre las capas adyacentes. La energía de corte para la base de onda plana establecida se estableció en 500 eV. El criterio de convergencia para la energía y la fuerza totales se estableció en 1 × 10 –6 eV y 0,01 eV / Å, respectivamente.
Resultados y discusiones
Energía de escisión, estado fundamental y estabilidad del MnBr 2 monocapa
Las constantes de celosía optimizadas de MnBr 2 a granel son a =b =3.95 Å, coherentes con el resultado experimental anterior ( a = b =3,87 Å) [25]. En primer lugar, exploramos la viabilidad de exfoliar MnBr 2 monocapa del volumen MnBr 2 . La Figura 1a presenta el método bien conocido, eficaz y ampliamente aprobado para calcular la energía de escisión [47, 48, 49]. Específicamente, la energía de escisión se obtuvo calculando la variación de la energía total del estado fundamental con respecto a la distancia de separación \ (d \) entre las dos partes de fractura como se muestra en la Fig. 1b, las constantes de celosía de ayb son fijado como los valores en el estado de equilibrio del volumen MnBr 2 . Las interacciones vdW de largo alcance entre capas se describieron mediante el esquema DFT-D2 de Grimme [50, 51]. La energía total aumenta con la distancia de separación y luego converge lentamente como se muestra en la Fig. 1b. La energía de escisión calculada es de 0,10 J / m 2 , que es más pequeña en comparación con la energía de escisión entre las dos partes de fractura del grafito (0,35 J / m 2 ) [52], que demuestra la viabilidad de obtener MnBr 2 monocapa mediante un método de exfoliación micromecánica.
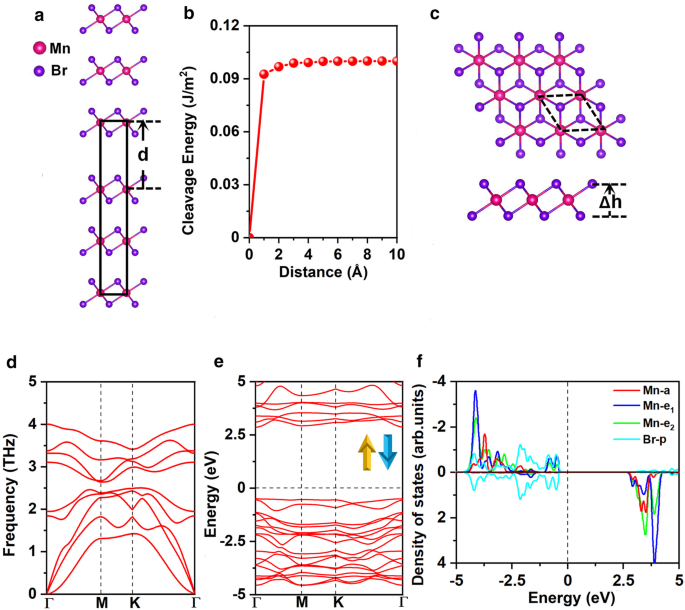
un Modelo masivo de MnBr 2 utilizado para calcular la energía de escisión y b la energía de escisión en función de la distancia de separación \ (d \) entre dos partes fracturadas (la distancia entre capas de equilibrio se establece en 0). c Vistas superior y lateral, d espectro telefónico, e estructura de banda electrónica para ambos canales de giro y f Densidad proyectada de estados (PDOS) de orbitales Mn-d y orbitales Br-p para MnBr 2 monocapa. Δ h representa la distancia vertical entre dos planos de haluro. La celda primitiva se distribuye en líneas de trazos negros. El nivel de Fermi para la estructura de banda y DOS se establece en 0 eV
MnBr 2 la monocapa tiene la simetría \ (C _ {{{3} v}} \) como se muestra en la Fig. 1c; cada átomo de Mn está rodeado por 6 átomos de Br vecinos, formando un octaédrico [MnBr 6 ] 4− unidad. Como se muestra en la Fig. 2a yb, se consideran tres posibles configuraciones magnéticas, a saber, estados no magnéticos (NM), ferromagnéticos (FM) y antiferromagnéticos (AFM). Se consideran los estados de alto y bajo giro del ion Mn. Nuestros resultados muestran que los iones Mn del estado FM están en bajo giro con d 1 configuración, mientras que los iones Mn en estado AFM están en alto giro con d 5 configuración. El estado fundamental de MnBr 2 monocapa es el estado AFM, que es más estable que los estados NM y FM en 3,91 eV y 0,72 eV por unidad de fórmula, respectivamente (archivo adicional 1:Tabla. S1). El MAE es de 0,25 meV, valor positivo que indica que el eje de magnetización fácil se encuentra a lo largo de las direcciones fuera del plano, coincidiendo con el resultado anterior [16]. Las constantes de celosía optimizadas son a = b =3.95 Å, lo mismo con las constantes de celosía del volumen MnBr 2 . La longitud del enlace Mn-Br es de 2,73 Å y la distancia vertical entre los dos planos de haluro es de 3,03 Å.
Diagramas esquemáticos para a ferromagnético y b configuraciones antiferromagnéticas para MnBr 2 monocapa
La estabilidad del MnBr 2 La monocapa se investigó más a fondo mediante el cálculo de la energía de formación, el espectro de fonones y las constantes elásticas. La energía de formación se calcula como:
$$ E _ {{{\ text {formulario}}}} =E _ {{{\ text {MnBr}} _ {{2}}}} - E _ {{{\ text {Mn}}}} - 2E _ {{ {\ text {Br}}}} $$donde \ (E _ {{{\ text {MnBr}} _ {{2}}}} \) representa la energía de MnBr 2 monocapa, \ (E _ {{{\ text {Mn}}}} \) y \ (E _ {{{\ text {Br}}}} \) son las energías de los átomos de Mn y Br en sus estructuras masivas, respectivamente. El \ (E _ {{{\ text {formulario}}}} \) calculado es - 1,87 eV por átomo; el valor negativo significa que la formación es exotérmica y MnBr 2 monocapa es energéticamente favorable. Además, nuestro espectro de fonones calculado (Fig. 1d) para MnBr 2 La monocapa no muestra una frecuencia negativa en toda la zona de Brillouin, lo que indica dinámicamente estable. Además, las constantes elásticas calculadas (Archivo adicional 1:Tabla S2) cumplen con los criterios de Born-Huang [53] de \ (C_ {11}> 0 \), \ (C_ {11} C_ {22} - C_ {12 } ^ {2}> 0 \) y \ (C_ {66}> 0 \), lo que confirma que MnBr 2 la monocapa es mecánicamente estable. La rigidez en el plano calculada es 26,98 J / m 2 , aproximadamente el 75% de MnPSe 3 (36 J / m 2 ) [49] y 15% de MoS 2 monocapa (180 J / m 2 ) [54]. Además, MnBr 2 La monocapa demuestra una mayor flexibilidad y la capacidad de soportar una mayor tensión de tracción en comparación con MoS 2 monocapa (11%) [54]. Esto puede atribuirse a enlaces iónicos para MnBr 2 monocapa contra los enlaces covalentes de MoS 2 monocapa. El análisis de la deformación relacionada con las constantes elásticas indica que puede soportar su peso (Ver detalles en el SI).
La estructura de banda electrónica de MnBr 2 La monocapa se muestra en la Fig. 1e, indica que MnBr 2 La monocapa es un semiconductor con una banda prohibida directa de 3,35 eV. Tanto el máximo de la banda de valencia (VBM) como el mínimo de la banda de conducción (CBM) están ubicados en el punto \ (\ Gamma \). Para comprender mejor las estructuras electrónicas, la densidad proyectada de estados (DOS) para el orbital Mn-d y Br-p se presenta en la Fig. 1f. Los cinco orbitales d del ion Mn se dividen en \ (a (d _ {{z ^ {2}}}) \), \ (e_ {1} (d_ {xz} + d_ {yz}) \) y \ ( e_ {2} (d_ {xy} + d _ {{x ^ {2} - y ^ {2}}}) \) grupos de acuerdo con la simetría \ (C _ {{{3} v}} \). El análisis de carga de bader sugiere que cada átomo de Mn dona dos electrones a los dos átomos de Br vecinos. Por lo tanto, los cinco orbitales d en un canal de espín están completamente ocupados por los cinco electrones d del Mn 2+ iones. En consecuencia, los dos Mn 2+ iones en la celda unitaria están en el d 5 estado de alto giro con el momento magnético de 5μ B / - 5μ B , el Br 1− Los iones están en el estado de giro bajo de 4p 6 con momento magnético despreciable de - 0.02μ B (Archivo adicional 1:Fig. S1 (a)). Según la regla de Goodenough-Kanamori-Anderson (GKA), dicha configuración siempre proporciona un acoplamiento antiferromagnético [55].
Estabilidad, propiedades electrónicas y magnéticas de Mn 3 Br 8 monocapa
Mn vacante se introdujo para romper el d 5 configuración del Mn 2+ iones. Se introduce una vacante de Mn único en la supercélula \ (2 \ times 2 \ times 1 \) de MnBr 2 monocapa, que emite el Mn 3 Br 8 monocapa. Como se muestra en la Fig. 3a, cada átomo de Mn tiene cuatro átomos de Mn vecinos más cercanos y se une a seis átomos de Br, formando un octaédrico distorsionado [MnBr 6 ] unidad. Se consideraron cinco estados magnéticos (NM, FM, FIM, AFM-1 y AFM-2) que se muestran en la Fig. 4. Nuestros resultados indican que el estado FM es el estado fundamental, que es más estable que los otros cuatro en 9,84 eV, 32,90 meV, 129,85 meV y 97,65 meV por unidad de fórmula, respectivamente. La constante de red optimizada sigue siendo 3,95 Å. Diferente de MnBr 2 monocapa, Mn 3 Br 8 La monocapa tiene 2 tipos de enlaces Mn-Br (Fig. 3b). Los enlaces entre el átomo de Mn y los dos átomos centrales de Br (\ (d _ {{\ text {Mn-Br1,2}}} \)) son 2,76 Å, mientras que los otros enlaces Mn – Br (\ (d _ {{\ text {Mn-Br3,4,5,6}}} \)) son 2,59 Å. La distancia vertical entre los dos planos de haluro es de 3,33 Å.
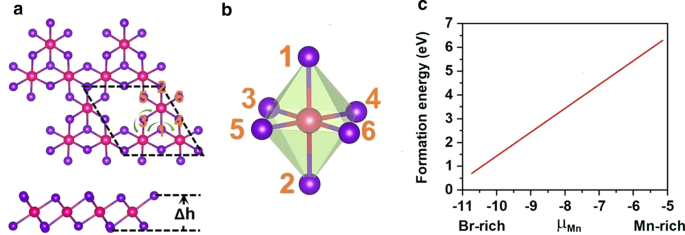
un Vistas superior y lateral de Mn 3 Br 8 monocapa, \ (\ Delta h \) representa la distancia vertical entre dos planos de haluro. La celda primitiva se distribuye en líneas de trazos negros; las líneas de flecha verde muestran dos caminos diferentes de la interacción de superintercambio. b Estructura del MnBr 6 distorsionado octaedro. c Energías de formación para una sola vacante de Mn en función del potencial químico de Mn (μMn)
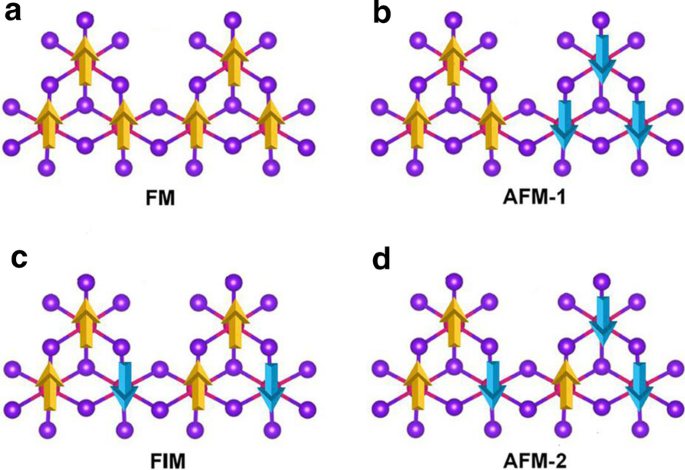
Diagramas esquemáticos para a ferromagnético, b antiferromagnético-1, c ferrimagnético y d configuraciones antiferromagnético-2 para Mn 3 Br 8 monocapa
Para verificar la viabilidad de inducir una vacante de Mn, en primer lugar calculamos las energías de formación de vacantes en entornos ricos en Mn y ricos en Br mediante las siguientes ecuaciones:
$$ E _ {{F ({\ text {Mn-rich}})}} {\ text {=}} E _ {{{\ text {Mn}} _ {3} {\ text {Br}} _ {8 }}} - (4 \ veces E _ {{{\ text {MnBr}} _ {{\ text {2}}}}} - \ mu _ {{{\ text {Mn-max}}}}) $$ $$ E _ {{F {\ text {(Br-rich)}}}} {=} E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8} }}} - (4 \ veces E _ {{{\ text {MnBr}} _ {{2}}}} - \ mu _ {{\ text {Mn-min}}}) $$donde \ (E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8}}}} \) y \ (E _ {{{\ text {MnBr}} _ {{2}}}} \) representan las energías totales del Mn 3 Br 8 y MnBr 2 monocapas, \ (\ mu _ {{\ text {Mn-max}}} \) es el potencial químico de Mn en un ambiente rico en Mn, que se calcula como la energía del átomo de Mn en su estructura global, \ (\ mu_ { {\ text {Mn-min}}} \) es el potencial químico de Mn en un entorno rico en Br, que se calcula como:
$$ \ mu _ {{\ text {Mn-min}}} =E _ {{{\ text {MnBr}} _ {{2}}}} - 2 \ times \ mu _ {{\ text {Br-max}} } $$donde \ (\ mu _ {{\ text {Br-max}}} \) es el potencial químico de Br y se calcula como la energía del átomo de Br en fase gaseosa. Como se muestra en la Fig. 3c, las energías de formación en un entorno rico en Mn / rico en Br son 6,30 / 0,71 eV por vacante de Mn, lo que indica que la formación de la vacante de Mn es energéticamente más favorable en el entorno rico en Br. De hecho, la vacante S se logró de manera experimental en MoS 2 monocapa [56], y la energía de formación predicha de la vacante S en el entorno rico en S es de 2,35 eV [57]. Además, la estructuración de nanoarquitectura porosa como β-FeOOH / PNGN (redes de grafeno poroso dopado con nitrógeno) puede inducir una vacante significativa de Fe [58], y se adoptó el método de Bridgman para inducir una vacante de Fe ordenada. También esperamos que estos métodos sean aplicables para inducir la vacante de Mn [59]. Además, no se encuentra una frecuencia negativa en el espectro de fonones de Mn 3 Br 8 monocapa que se muestra en la Fig. 5a, lo que demuestra la estabilidad dinámica. Estos resultados aprueban nuestro diseño de introducir Mn vacante para traer ferromagnetismo.
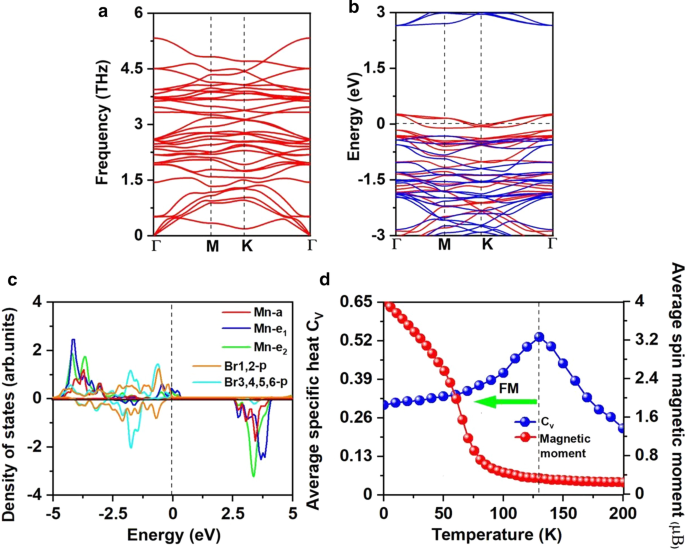
un Espectros de fonones, b estructura de banda electrónica resuelta por giro, y c Densidad proyectada de estados (PDOS) de orbitales Mn-d y orbitales Br-p para Mn 3 Br 8 monocapa. d Momentos magnéticos in situ de los átomos de Mn y el calor específico C v en función de la temperatura según el modelo de Heisenberg para Mn 3 Br 8 monocapa. El nivel de Fermi para la estructura de banda y PDOS se establece en 0 eV
El ferromagnetismo de Mn 3 Br 8 atributos de monocapa a la interacción de superintercambio FM. Según la regla de Goodenough-Kanamori-Anderson (GKA) [55], la interacción de superintercambio entre los iones Mn es FM cuando el ángulo Mn-Br-Mn es de alrededor de 90 °. En dicha configuración (archivo adicional 1:Fig. S2), el orbital Mn-d tiende a acoplarse AFM con diferentes orbitales Br-p ortogonales y, por lo tanto, se espera que el acoplamiento magnético indirecto Mn-Mn sea FM. Pero si cada ion Mn tiene 5 electrones desapareados como MnBr 2 monocapa, el superintercambio es AFM, aunque el ángulo Mn-Br-Mn está cerca de 90 ° porque no quedan orbitales Mn-d de spin-up vacíos en MnBr 2 Los electrones d monocapa y spin-up no pueden saltar entre el sitio Mn vecino [60]. Existen dos rutas de interacción de superintercambio diferentes en Mn 3 Br 8 (Fig. 3a), y ambos son FM. Uno involucra átomos centrales Br1,2 con longitudes de enlace Mn-Br de 2,76 Å y ángulos Mn-Br-Mn de 87,5 °; el otro involucra átomos de Br3,4,5,6 con una longitud de enlace Mn-Br de 2.59 Å y ángulos Mn-Br-Mn de 95 °. Las interacciones hibridadas entre los orbitales p de los átomos Br3,4,5,6 y los orbitales Mn-d son más fuertes que las de la hibridación p-d que involucra átomos Br1,2, como se muestra en la Fig. 5c, particularmente de -2 eV a -1,4 eV. Mientras que de 1,4 a - 0,9 eV, la p - d la hibridación que involucra átomos de Br1,2 está dominada.
El análisis de carga de bader sugiere que cada átomo de Mn dona 8/3 electrones a los átomos de Br vecinos. Por lo tanto, los iones Mn están en el Mn 8/3 + Expresar. Como se muestra en la Fig. 5c, los 13/3 electrones de cada ion Mn llenan el canal de giro del orbital d, mientras que el Br 1− Los iones están en el estado de giro bajo de 4p 6 . Por lo tanto, el momento magnético de cada Mn 8/3 + ion es 13 / 3μ B ; el momento magnético de Br 1− los iones son despreciables (archivo adicional 1:Fig. S1 (b)). La inducción de ferromagnetismo por vacante también se puede observar para el d 0 sistemas, como ZnS y ZnO [61, 62], una sola vacante puede inducir un momento magnético tan grande como 2μ B [61] . Por cada ion Mn, 2/3 d-orbital están desocupados; el canal de giro de los orbitales \ (e_ {1} \) y \ (e _ {{2}} \) están parcialmente ocupados y cruzan el nivel de Fermi, lo que resulta en una semimetalicidad. El carácter semimetálico también se puede observar en la estructura de banda electrónica resuelta por espín que se muestra en la Fig. 5b. El canal de giro es metálico, mientras que el canal de giro es semiconductor con un intervalo de banda indirecto de 2,97 eV; el VBM / CBM se ubica en el punto \ ({\ text {M}} \) / \ (\ Gamma \). El valor de la banda prohibida es cercano a los de MnP (2,86 eV) [63], MnAs (2,92 eV) [63] y Ni 2 NO 2 (2,98 eV) [64], que es lo suficientemente grande como para evitar el giro de giro excitado térmicamente. Comparando con el MnBr 2 monocapa, tanto el VBM como el CBM del canal semiconductor se acercan más al nivel de Fermi. El CBM todavía está dominado por los átomos de Mn, mientras que el VBM está dominado por los nuevos átomos Br1,2. Mientras tanto, el canal semiconductor se convierte de directo a indirecto y la banda prohibida se reduce. Se observó un fenómeno similar en MnCl 2 monocapa con funcionalización H [60].
Las direcciones de magnetización están determinadas por la energía de anisotropía magnética (MAE). El MAE de los sólidos surge de dos contribuyentes, a saber, la energía magnetocristalina (MCE) relacionada con el acoplamiento espín-órbita (SOC) y la energía de anisotropía dipolar magnética (MDE) atribuida por la interacción magneto-estática dipolo-dipolo. El MDE en los materiales isotrópicos 3D, como bcc Fe y fcc Ni, es muy pequeño. Pero para materiales de baja dimensión compuestos por átomos de metales de transición con gran momento magnético, el MDE no debe ignorarse [65,66,67]. El MCE se define como la diferencia entre la energía de magnetización a lo largo de las direcciones en el plano (100 o 010) y fuera del plano (001) teniendo en cuenta el SOC. El MDE se obtiene como la diferencia de \ (E_ {d} \) entre las magnetizaciones en el plano y fuera del plano. \ (E_ {d} \) en unidades atómicas de Rydberg viene dado por [65, 66]
$$ E_ {d} =\ sum \ limits_ {ij} {\ frac {{2m_ {i} m_ {j}}} {{c ^ {2}}}} M_ {ij} $$donde la velocidad de la luz, \ (c =274.072 \), \ (i / j \) son los vectores de posición atómica en la celda unitaria, y \ ({m} _ {i} / {m} _ {j} \ ) es el momento magnético atómico (μ B ) en el sitio \ (i / j \). La constante magnética dipolar de Madelung \ (M_ {ij} \) se calcula mediante
$$ M_ {ij} =\ sum \ limits_ {R} {\ frac {1} {{\ left | {R + i + j} \ right | ^ {3}}}} \ left \ {{1 - 3 \ left. {\ frac {{\ left [{(R + i + j) \ cdot \ mathop {m_ {i}} \ limits ^ {\ wedge}} \ right] ^ {2}}} {{\ left | {R + i + j} \ right | ^ {2}}}} \ right \}} \ right. $$donde \ (R \) son los vectores reticulares. En un material 2D, dado que todos los \ (R \) y \ (i \) están en el plano, el segundo término sería cero para la magnetización fuera del plano, lo que resulta en \ (M_ {ij} \ ), mientras que \ (M_ {ij} \) es negativo para una magnetización en el plano [67]. Por lo tanto, el MDE se relaciona con el momento magnético del metal de transición y siempre prefiere la magnetización en el plano.
El MCE calculado para Mn 3 Br 8 La monocapa es - 1.90 meV por unidad de fórmula (Fig. 6a), mucho más grande que las de Fe a granel (0.001 meV por átomo) y Ni (0.003 meV por átomo) [68], y más grande que la de la monocapa de Fe en Rh (111) (0.08 meV por átomo) [69], lo que sugiere que la magnetización del Mn 3 Br 8 La monocapa es termoestable. La relación entre el MCE y el ángulo azimutal puede describirse mediante la siguiente ecuación [70]:
$$ {\ text {MCE}} (\ theta) =A \ cos ^ {2} (\ theta) + B \ cos ^ {4} (\ theta) $$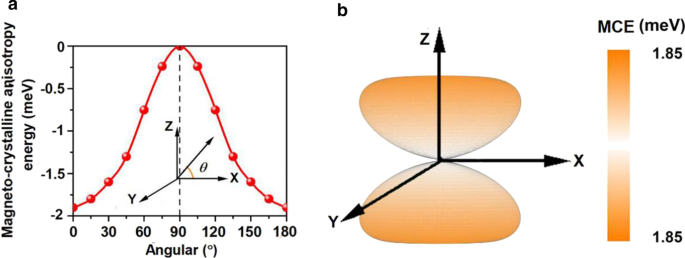
Variación de la energía de anisotropía magnetocristalina (MCE) a con respecto al ángulo azimutal y b en el espacio para Mn 3 Br 8 monocapa
donde \ (A \) y \ (B \) son las constantes de anisotropía y \ (\ theta \) es el ángulo azimutal. El resultado del ajuste se muestra en el archivo adicional 1:Figs. S3. Además, la evolución de MCE con el eje de giro girando a través de todo el espacio se ilustra en la Fig. 6b. MCE dentro del plano xy no muestra diferencia, pero alcanza el valor máximo a lo largo de la dirección perpendicular al plano xy, lo que confirma la fuerte anisotropía magnética. El MDE es - 0.43 meV por unidad de fórmula y MAE (MCE + MDE) es - 2.33 meV por unidad de fórmula. El valor negativo indica que el eje de magnetización fácil se encuentra a lo largo de las direcciones en el plano. El MDE no cambia la dirección de magnetización, pero la mejora. Además, el MAE de Mn 3 Br 8 la monocapa es mucho más grande que la de MnBr 2 monocapa, demostrando una vez más la eficacia de nuestro diseño.
Calculamos además el \ (T_ {c} \) para FM Mn 3 Br 8 monocapa mediante la realización de simulaciones de Monte Carlo (MC) basadas en el modelo de Heisenberg, que ha demostrado ser el método eficaz para predecir \ (T_ {c} \) para materiales 2D [11, 15, 48, 58, 71,72 , 73,74,75,76]. Nuestro \ (T_ {c} \) estimado de CrI 3 monocapa es 42 K (archivo adicional 1:Fig. S4) [76], concordando bien con el valor medido experimental [2] y los resultados de cálculos anteriores [15, 58, 71, 72, 74, 76], lo que demuestra la precisión de nuestro método adoptado. El spin-hamiltoniano que incluye la interacción magnética vecina más cercana (NN) se describe como
$$ H =- \ sum \ limits_ {i, j} {JM_ {i} M_ {j}} $$donde \ (J \) es el parámetro de intercambio magnético NN, \ (M_ {i / j} \) es el momento magnético de los iones Mn y la integral cercana al número de electrones polarizados de espín según el método de Monte Carlo [71, 77 , 78], \ (i \) y \ (j \) representan el par NN de iones Mn. El parámetro de acoplamiento magnético \ (J \) se calcula mediante la diferencia de energía entre los estados FM y AFM como
$$ J {=} \ frac {{E _ {{{\ text {AFM1}}}} - E _ {{{\ text {FM}}}}}} {{16M ^ {2}}} $$El \ (J \) calculado de NN Mn iones es 1.01 meV; el valor positivo indica la preferencia del acoplamiento FM.
El \ (J \) calculado de los iones NN Mn y la supercélula \ (100 \ times 100 \ times 1 \) que contiene 20.000 vectores de momento magnético se adoptaron para realizar las simulaciones de MC. Las simulaciones a cada temperatura tienen una duración de 10 5 pasos. Cada vector de momento magnético gira aleatoriamente en todas las direcciones. La Figura 5d muestra la evolución del calor específico definido como \ (C _ {{_ {V}}} ={{\ left ({\ left \ langle {E ^ {2}} \ right \ rangle - \ left \ langle E \ right \ rangle ^ {2}} \ right)} \ mathord {\ left / {\ vphantom {{\ left ({\ left \ langle {E ^ {2}} \ right \ rangle - \ left \ langle E \ right \ rangle ^ {2}} \ right)} {K_ {B} T ^ {2}}}} \ right. \ kern- \ nulldelimiterspace} {K_ {B} T ^ {2}}} \) con temperatura, de donde obtuvimos el \ (T_ {c} \) de 130 K para Mn 3 Br 8 monocapa al ubicar la posición máxima de \ (C_ {v} \), más alta que la temperatura del nitrógeno líquido (77 K), y \ (T_ {c} \) de CrI 3 (45 K) [2] y Cr 2 Ge 2 Te 6 (28 K) [3], CrX 3 (X =F, Cl, Br) (36 ~ 51 K) [11], CrXTe 3 (X =Si, Ge) (35,7 K, 57,2 K) [48]. Nuestros cálculos demuestran que el FM Mn 3 Br 8 La monocapa tiene la temperatura MAE grande y Curie más alta que la temperatura del nitrógeno líquido.
Mn 3 Br 8 monocapa bajo tensión biaxial y dopaje con portador
Se ha demostrado que la ingeniería de deformación es aplicable para muchos materiales 2D y eficaz para alterar los parámetros estructurales, como las longitudes y ángulos de unión, y ajustar las propiedades electrónicas y magnéticas. En este contexto, investigamos Mn 3 Br 8 monocapa bajo la tensión biaxial que varía de - 5% a 5%. Resulta que Mn 3 Br 8 La monocapa bajo tensión biaxial de - 5 a 5% se mantiene FM y el momento magnético atómico apenas cambia. Como se muestra en las Figs. 7a yc, los ángulos entre dos átomos de Mn y átomos de Br1,2 (θ Mn-Br1,2-Mn ) son 84 ° -90 °, que aumentan a medida que la deformación y se acerca gradualmente a 90 °. Los ángulos Mn-Br-Mn que involucran Br3,4,5,6 átomos (θ Mn-Br3,4,5,6-Mn ) se desvían gradualmente de 90 °, oscilando entre 90 ° y 100 °. Por lo tanto, las interacciones de superintercambio entre los iones Mn mediadas a través de diferentes orbitales Br-p ortogonales siguen siendo FM.
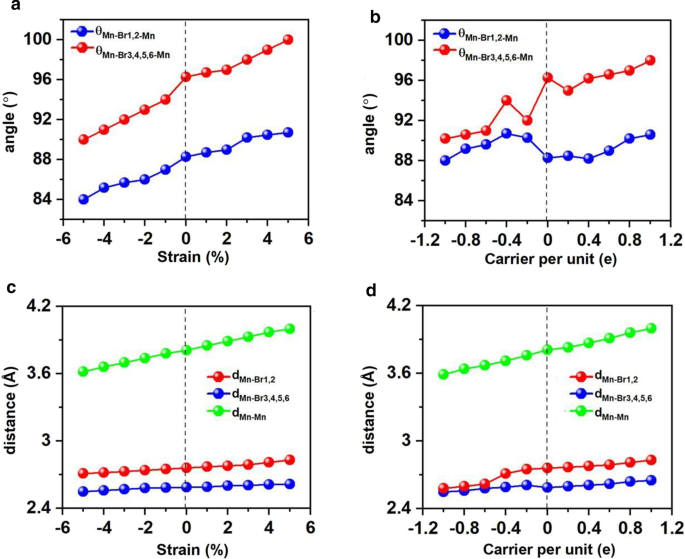
Variaciones de los ángulos entre dos átomos de Mn y Br, la distancia entre los átomos de Mn y Br y la distancia entre los átomos de Mn vecinos más cercanos con respecto a la deformación biaxial aplicada y al dopaje del portador. Variación de a ángulo y c distancia con respecto a la deformación biaxial, variaciones de b ángulo y d distancia con respecto al dopaje por portador. Los valores positivos y negativos del dopaje de portadores representan el dopaje de electrones y huecos, respectivamente
Tanto las distancias Mn-Mn como Mn-Br aumentan monótonamente a medida que la deformación cambia de –5% a 5%. En consecuencia, el parámetro de intercambio bajo la deformación biaxial que se presenta en la Fig. 8a disminuye con la deformación biaxial cambiando de –5% a 5% y alcanza el valor más grande (1,18 meV) bajo una deformación biaxial de –5%. La temperatura de Curie de Mn 3 Br 8 la monocapa con una deformación biaxial de –5% es de 160 K (Fig. 9a). Particularly, the Mn-Br bonds under the increasing tensile strain become longer, and the angles of Mn-Br3,4,5,6-Mn deviate from 90°, which are the main reason why the FM super-exchange interaction becomes weaker. Consequently, the Curie temperature decreases. It is similar with CrPTe3 and FePS3 monolayers [79]. Additionally, the MDE decreases with the increasing strain (Additional file 1:Fig. S5(b)); the MAE under –1% biaxial strain is the largest (–3.04 meV). The –5–5% strain does not cause large structural deformation for Mn3 Br8 monolayer, and the morphology of its band structures hardly changes. Mn3 Br8 monolayer keeps to be half-metallic. Both VBM and CBM in the semiconducting spin-channel move upward slightly to the higher energy as shown in Figs. 8c and 10; the band gap increases slowly with the increasing biaxial strain to 3.12 eV under 5% biaxial strain.

Variations of a the exchange parameter and b magnetic anisotropy energy (MAE) for Mn3 Br8 monolayer with respect to the applied biaxial strain and carrier doping. The variations of valence band maximum (VBM), conduction band minimum (CBM), and band gap in the semiconducting channel for Mn3 Br8 monolayer with respect to c the applied biaxial strain and d carrier doping ranging. Positive and negative values of the carrier doping represent the electron and hole doping, respectively

On-site magnetic moments of Mn atoms and the specific heat C v as function of temperature based on Heisenberg model for Mn3 Br8 monolayer a under -5% biaxial strain, with b 0.2e, c -0.6e, and d -0.8e carrier doping per formula unit. Positive and negative values represent the electron and hole doping, respectively

un - j Spin-resolved band structure for Mn3 Br8 monolayer under biaxial strain from -5% to 5%. The green arrow denotes the indirect band gap
Electron/hole doping always leads to VBM/CBM moving away from the Fermi level. Our calculations show that Mn3 Br8 monolayer with –1–1e (~ \(1.7 \times 10^{14} {\text{cm}}^{{ - 2}}\)) carrier doping per formula unit is still FM; the atomic magnetic moment of each Mn ion is still 13/3μB. As shown in Fig. 7b and d, with carrier doping from –1e to 1e per formula unit, the Mn-Br-Mn angles involving Br3,4,5,6 atoms are about 90° ~ 98°; the Mn-Br1,2-Mn angles are about 88° ~ 90°. The Mn–Mn and Mn-Br1,2 distances increase with the increasing electron doping. Mn3 Br8 monolayer with 0.2e and 0.4e carrier doping has larger magnetic exchange parameter (Fig. 8a). The Curie temperature at 0.2e electron doping is largest of 140 K (Fig. 9b). Additionally, with –1e ~ 0.2e doping, the MAE is along in-plane directions; the MDE decreases with the increasing electron doping. Under 0.4e doping, the MCE turns to be positive with the value of 0.41 meV per formula unit; the MAE is only 0.01 meV per formula unit with taking the MDE into account (Additional file 1:Figs. S5(a) and (b)). With 0.6e, 0.8e and 1e doping, the PMA (perpendicular magnetic anisotropy energy) is 1.70, 2.42, and 5.13 meV, respectively, large enough for spintronic applications (Fig. 8b).
Additionally, Mn3 Br8 monolayer with carrier doping of –1e ~ 1e per formula unit maintains to be half-metallic. Its band gap in the semiconducting spin-channel increases/decreases slightly with the increasing electron/hole doping as shown in Fig. 8d; the positions of the VBM and CBM do not change. Exceptional, Mn3 Br8 monolayer turns to be FM spin-gapless semiconductors (SGS) with the metallic spin-channel opening up a very small energy gap (0.07 eV) under –0.6e and –0.8e hole doping; its Fermi level locates in the band gap region (Fig. 11b and c, more clearly figures are presented in Additional file 1:Figs. S6(a) and (b)). Correspondingly, electrons may be easily excited from the valence band to the conduction band with a small input of energy, which simultaneously produces 100% spin polarized electron and hole carriers. The Curie temperature at –0.6e and –0.8e hole doping is 110 K (Fig. 9c and d), higher than liquid-nitrogen temperature (77 K). Considering with that the charge density modulation of \(10^{13} \sim10^{15} {\text{cm}}^{ - 2}\) was already achieved experimentally [80,81,82], our predicted properties of Mn3 Br8 monolayer with carrier doping are also experimentally approachable.

un - j Spin-resolved band structure for Mn3 Br8 monolayer with carrier doping from -1e to 1e per formula unit. Positive and negative values represent the electron and hole doping, respectively. The green arrow denotes the indirect band gap
Conclusiones
In summary, the stability, electronic, and magnetic properties of Mn3 Br8 monolayer have been carefully investigated. Our results show that Mn3 Br8 monolayer is FM half-metal with 130 K Curie temperature and with 2.97 eV band gap for the semiconducting spin-channel. Plus, the magnetic moment of each Mn ion is 13/3μB; the MAE is –2.33 meV per formula unit. The Mn3 Br8 monolayer is designed by inducing single Mn vacancy in the \({2} \times {2} \times {1}\) supercell of MnBr2 monolayer to break the AFM coupling d 5 configuración. The feasibility of forming the Mn vacancy and the dynamical, mechanical stability of Mn3 Br8 monolayer have been comprehensively confirmed. Additionally, Mn3 Br8 monolayer under biaxial strain –5% ~ 5% is still FM half-metal with 2.71 ~ 3.12 eV band gap for the semiconducting spin-channel, whose Curie temperature under –5% biaxial strain is 160 K. Both biaxial strain and carrier doping make the MAE increase, which turns to be perpendicular to the plane under electron doping. With 0.8e and 0.6e hole doping, Mn3 Br8 monolayer turns to be spin-gapless semiconductor (SGS) with band gap of 0.07 eV. Our calculations demonstrate Mn3 Br8 monolayer as FM half-metal with high Curie temperature, and having large MAE and large magnetic moment, and tunable electronic and magnetic properties under the applied biaxial strain and carrier doping.
Disponibilidad de datos y materiales
Los conjuntos de datos generados durante y / o analizados durante el estudio actual están disponibles del autor correspondiente a solicitud razonable.
Abreviaturas
- 2D:
-
Bidimensional
- AFM:
-
Antiferromagnetic
- CBM:
-
Banda de conducción mínima
- DFT:
-
Teoría funcional de la densidad
- DOS:
-
Densidad de estados
- FIM:
-
Ferrimagnetic
- FM:
-
Ferromagnetic
- GGA:
-
Aproximación de gradiente generalizada
- GKA:
-
Goodenough–Kanamori–Anderson
- MAE:
-
Magnetic anisotropy energy
- MCE:
-
Magneto-crystalline anisotropy energy
- MC:
-
Monte Carlo
- MDE:
-
Magnetic dipolar anisotropy energy
- MTJ:
-
Magnetic tunneling junctions
- NM:
-
Non-magnetic
- NN:
-
Nearest neighboring
- PAW:
-
Proyector de onda aumentada
- PBE:
-
Perdew–Burke–Ernzerhof
- PMA:
-
Perpendicular magnetic anisotropy energy
- PNGN:
-
Porous nitrogen-doped graphene networks
- SGS:
-
Spin-gapless semiconductor
- SOC:
-
Spin–orbit coupling
- VASP:
-
Vienna ab-initio simulation package
- VBM:
-
Máximo de banda de valencia
- VDW:
-
Van der Waals
Nanomateriales
- Entrevista con Craig Trevor de Persuasion Inc.
- De la implementación piloto a la implementación a gran escala:Llegando a la distancia con IoT
- La bifurcación de susceptibilidad magnética en el aislante topológico Sb2Te3 dopado con Ni con orden antiferromagnético acompañado de una alineación ferromagnética débil
- Emisión multicolor de una estructura de nanopiramida cuasicristalina fotónica ultravioleta basada en GaN con múltiples pozos cuánticos semipolares InxGa1 − xN / GaN
- Absorbedor perfecto de banda ancha con monocapa MoS2 y matriz de nanodiscos de nitruro de titanio hexagonal
- Microesferas de carbono magnéticas como adsorbente reutilizable para la eliminación de sulfonamidas del agua
- Respuesta fotovoltaica pronunciada del fototransistor MoTe2 multicapa con formulario de contacto asimétrico
- Evolución del área de contacto con carga normal para superficies rugosas:de escalas atómicas a macroscópicas
- Mejora de la absorción de banda ancha y multibanda de grafeno monocapa en frecuencias ópticas a partir de resonancias magnéticas dipolo múltiples en metamateriales
- Eliminación de antibióticos del agua con una membrana de nanofiltración 3D totalmente de carbono
- Histéresis magnética en nanoestructuras con acoplamiento RKKY controlado térmicamente