


Sensor químico de etanol altamente sensible basado en el novedoso α-Fe2O3 mesoporoso dopado con ag preparado mediante el proceso de sol-gel modificado
Resumen
Α – Fe 2 mesoporoso O 3 se ha sintetizado mediante un sencillo procedimiento sol-gel en presencia de copolímero tribloque Pluronic (F-127) como agente director de estructura. Se depositaron nanopartículas de plata (Ag) en α – Fe 2 O 3 matriz mediante el enfoque de reducción fotoquímica. El análisis morfológico reveló la formación de nanopartículas de Ag con tamaños pequeños <20 nm en la estructura mesoporosa de α – Fe 2 O 3 que posee una forma semiesférica de <50 nm. XRD, FTIR, Raman, UV-vis, PL y N 2 Los estudios de isotermas de sorción confirmaron la alta cristalinidad, mesoporosidad y características ópticas del producto sintetizado. La detección electroquímica hacia el etanol líquido se ha realizado utilizando la corriente descentrada Ag / α – Fe 2 O 3 -Electrodo de carbono vítreo modificado (GCE) por voltamperometría cíclica ( CV ) y potencial actual ( I-V ) técnicas, y los resultados obtenidos se compararon con GCE desnudo o α-Fe 2 puro O 3 . Mesoporosa Ag / α – Fe 2 O 3 Se descubrió que mejoraba en gran medida la sensibilidad del sensor y exhibía excelentes características de detección durante la detección de precisión de bajas concentraciones de etanol. Sensibilidad alta y reproducible de 41,27 μAmM - 1 cm - 2 en la región de menor concentración de etanol (0,05 a 0,8 mM) y 2,93 μAmM - 1 cm - 2 en la zona de mayor concentración (0,8 a 15 mM), con un límite de detección (LOD) de 15,4 μM. La investigación sobre la cinética de reacción reveló un comportamiento característico de la superficie mixta y los procesos controlados por difusión. Los estudios de detección detallados revelaron también que la sensibilidad hacia el etanol era más alta que la del metanol o el isopropanol. Con un mayor esfuerzo en el desarrollo de los enfoques de síntesis y fabricación, es posible una utilidad adecuada para el protocolo propuesto actual para fabricar un mejor rendimiento del dispositivo sensor.
Antecedentes
El área de investigación de los sensores químicos se ha expandido significativamente en la última década debido a su importancia en una amplia gama de aplicaciones tecnológicas en los campos del diagnóstico y descubrimiento de fármacos, temas relacionados con la seguridad, industrias alimentarias, monitoreo ambiental y análisis agrícolas [1, 2]. Con base en la propiedad física por determinar, los sensores químicos podrían clasificarse como sensores ópticos, eléctricos, térmicos o de masa, y son apropiados para detectar analitos objetivo en estado gaseoso, líquido o sólido [3]. Entre los sensores actualmente disponibles, los sensores electroquímicos son particularmente atractivos debido a su notable sensibilidad, tiempo de respuesta rápido esperado, simplicidad de configuración experimental y menor costo [4]. En los sensores electroquímicos, los electrodos de trabajo se modifican esencialmente con los materiales de detección activos. Las propiedades físico-químicas de los materiales activos afectan en gran medida al rendimiento del sensor, así como a su estabilidad operativa [5]. Por lo tanto, la investigación y el desarrollo de un material activo potencial juega un papel decisivo en la fabricación de dispositivos de detección sensibles, eficientes y confiables. Además, con la ayuda de la nanotecnología, ahora es probable que sintetice una amplia gama de nuevos nanomateriales con formas y morfologías específicas, lo que podría dar lugar a características físico-químicas únicas [6, 7, 8]. Particularmente, los semiconductores de óxido metálico son una clase única de nanomateriales que han recibido considerable atención debido a sus prometedores rendimientos de detección, ya que podrían promover la cinética de transferencia de electrones [9,10,11,12,13], además de su atractivo características tales como facilidad de fabricación, capacidad para controlar el tamaño y la morfología, facilidad para modificar la superficie, buena estabilidad química y propiedades catalíticas [14]. También mostraron una fuerte afinidad hacia la adsorción de moléculas diana [15,16,17,18]. Se han sintetizado con éxito varios tipos de semiconductores de óxido metálico con diferentes morfologías; nanopartículas, nanocables, nanobarras, nanotubos, nanohojas, nanocinturones y puntos cuánticos utilizando diversas rutas sintéticas como hidrotermal / solvotermal [19,20,21], sol-gel [22, 23], crecimiento en soluciones acuosas [24], química deposición [25], técnica electroquímica [26] y deposición química y física de vapor [27, 28]. Sin embargo, el desarrollo de semiconductores de óxido metálico novedosos y efectivos para aplicaciones de sensores químicos sigue siendo un desafío existente que requiere una manipulación adecuada y optimización de materiales con una selección cuidadosa del electrodo de trabajo apropiado.
Como n semiconductor de tipo α – Fe 2 O 3 (fase hematita de los óxidos de hierro) es una categoría de óxido notablemente prometedora que se caracteriza por su alta estabilidad, resistencia a la corrosión, no toxicidad, y ha encontrado un amplio uso como material de detección de gases y químicos [29,30,31], como pigmentos y en medios de grabación magnéticos , fotocatálisis y fotoanodo en desdoblamiento de agua [32,33,34]. Por ejemplo, sensor químico basado en α – Fe 2 O 3 Se han fabricado nanopartículas con una variación de alta resistencia para la detección de CH 3 Gas SH, a temperatura ambiente en el rango de 20 a 80 ppm [35]. En otro informe, Fe 2 dopado con Ag O 3 ya que los nanocompuestos core-shell han mostrado una buena sensibilidad al NO 2 gas y podría detectar tan solo 0,5 ppm de NO 2 [36]. Un nanocompuesto terciario de Ag – Fe 2 O 3 –RGO también se sintetizó mediante reducción química y método hidrotermal y se empleó con éxito como un H 2 no enzimático O 2 sensor [37]. Un nanocompuesto de α – Fe 2 O 3 –GO con Fe 2 diferente O 3 Los contenidos se han diseñado y utilizado para mejorar el rendimiento de detección del gas etanol [38]. En esta contribución, un nuevo Ag / α – Fe 2 O 3 La nanoestructura híbrida se ha sintetizado a través de un procedimiento sol-gel simple y modificado utilizando el copolímero tribloque Pluronic (F-127) como agente director de estructura seguido de un enfoque de fotorreducción para depositar nanopartículas de Ag. El mesoporoso recién desarrollado Ag / α – Fe 2 O 3 Se han explorado las atractivas propiedades de ambos componentes (nanopartículas de metales nobles y óxido de metal mesoporoso) como un sensor químico sensible para detectar eficazmente etanol líquido a baja concentración mediante voltamperometría cíclica y potencial de corriente ( I-V ) técnicas. Hasta donde sabemos, la mesoestructura híbrida propuesta actualmente no se ha utilizado antes para la detección electroquímica de etanol.
Métodos / Experimental
Materiales
El tensioactivo de copolímero de bloque EO 106 –PO 70 EO 106 (F-127, EO = –CH 2 CH 2 O–, PO = –CH 2 (CH 3 ) CHO–), PM 12600 g / mol), nitrato de hierro Fe (NO 3 ) 3 .9H 2 O, etanol C 2 H 5 OH, nitrato de plata AgNO 3 se compraron a Sigma-Aldrich y se usaron tal como se recibieron sin purificación adicional.
Síntesis de α – Fe 2 mesoporoso O 3
Α – Fe 2 mesoporoso O 3 Los nanocristales se sintetizaron mediante un procedimiento de sol-gel utilizando F-127 como agente director de molde. Se emplearon las siguientes relaciones molares de precursores de partida:Fe (NO 3 ) 3 .9H 2 O / F127 / C 2 H 5 OH / HCl / CH 3 COOH =1:0,02:50:2,25:3,75. En una corrida sintética típica, se añadieron 1,6 g de F127 a 30 ml de etanol con agitación hasta obtener una solución transparente. Luego, 2,3 ml de CH 3 Posteriormente se añadieron COOH, 0,74 ml de HCl y 4,4 g de nitrato de hierro a la solución anterior con agitación vigorosa durante 60 min y finalmente se transfirieron a una placa de Petri para la etapa de gelificación. La mesofase sintetizada se secó y envejeció a 40 ° C y 40% de humedad durante 12 h, seguido de un envejecimiento adicional a 65 ° C durante 24 h. Se realizó un paso de calcinación y se adaptó a 450 ° C durante 4 h a una velocidad de calentamiento de 1 ° C / min y una velocidad de enfriamiento de 1 ° C / min para obtener α-Fe 2 mesoporosa O 3 nanocristales.
Reducción fotoquímica de iones Ag en mesoporosa α – Fe 2 O 3
Ag se depositó en α-Fe 2 mesoporoso O 3 mediante la reducción fotoquímica de iones de plata de acuerdo con el siguiente procedimiento:una solución en suspensión que contiene 1,0 g de α-Fe 2 mesoporoso O 3 y 9,4 × 10 - 5 mol AgNO 3 se sonicó en 100 ml de metanol acuoso (1% ( v / v ) metanol / H 2 O). La solución se iluminó con una lámpara de Philips Hg con luz UV (A) (intensidad =2,0 mWcm - 2 ) durante 12 h. El Ag / α – Fe 2 producido O 3 se separó por centrifugación, se lavó con agua desionizada y etanol y se secó a 110 ° C durante 12 h.
Caracterización de materiales
Los patrones de difracción de rayos X (XRD) se midieron con un difractómetro de puerto PANalytical X 'usando Cu Kα 1/2 , λα 1 =154.060 pm, λα 2 =154.439 pm de radiación. El espectro del espectrómetro infrarrojo de transformadas de Fourier (FT-IR) se recopiló en el rango de 400 a 4000 cm - 1 utilizando el espectrómetro BRUKER FRA 106 utilizando el procedimiento estándar de pellets de KBr. Los espectros Raman se midieron usando una estación Raman 400 de Perkin Elmer. Se usó un espectrofotómetro UV-visible (lambda 950 Perkin Elmer) para la medición de los espectros de absorción óptica UV-vis a temperatura ambiente en el rango de 200-800 nm. Los espectros de fotoluminiscencia (PL) a temperatura ambiente se recogieron en un espectrofluorofotómetro (RF-5301 PC, Japón, SHIMADZU, 400 W, 50/60 Hz) usando una lámpara de xenón de 150 W a una longitud de onda de excitación de 315 nm. La morfología de la superficie se investigó mediante microscopio electrónico secundario de emisión de campo (FE-SEM) con un microanalizador electrónico de barrido FE (JEOL-6300F, 5 kV), equipado con análisis EDS. La estación A de Quantachrome NOVA se utilizó para obtener isotermas de adsorción / desorción de nitrógeno a 77 K para las muestras secadas al vacío a 300 ° C durante 3 h. Se aplicó el modelo de Barrett-Joyner-Halenda (BJH) con la ecuación de Halsey para calcular los datos de sorción [39].
Detección electroquímica de etanol en soluciones acuosas
Electrodos de carbono vítreo (GCE) con una superficie de 0,071 cm 2 (Bio-Logic SAS) se pulieron inicialmente con 1 y 0,05 μm de pulido de diamante y lechada de alúmina, respectivamente, se lavaron con agua desionizada, se sonicaron en etanol, agua y finalmente se dejaron secar de forma natural. El GCE fue posteriormente recubierto por Ag / α – Fe 2 O 3 material activo usando un acetato de butilcarbitol y acetato de etilo como aglutinantes conductores. A continuación, la GCE modificada se secó durante la noche a 65ºC. Se conectó una celda electroquímica típica de dos electrodos con un electrodo de trabajo (GCE modificado) y un contraelectrodo (un cable de Pt) a la estación de trabajo electroquímica, ZahnerZennium, Alemania. También se utilizó una celda de tres electrodos que usa un electrodo de referencia de Ag / AgCl para la investigación de voltamperometría cíclica. Se preparó una concentración 0,1 M de PBS (solución tampón de fosfato) de pH 7 a partir de Na 2 HPO 4 y NaH 2 PO 4 y actuó como electrolito de soporte. En este estudio se aplicaron varias concentraciones de etanol que van desde 0,05 a 15 mM. El I-V Las características (corriente-potencial) se midieron con agitación continua, a temperatura ambiente, en la dirección anódica dentro de una ventana de potencial de 0 a 1,5 V a una velocidad de exploración de 50 mV / s. La sensibilidad del sensor se estimó a partir de la pendiente de la curva de calibración correspondiente de la concentración de corriente frente a la de etanol dividida por el área de superficie de GCE. El LOD (límite de detección) se calculó en una S / N =3 (relación señal / ruido). Una ilustración esquemática para la síntesis de Ag / α – Fe 2 O 3 con la detección electroquímica de etanol se muestra en el Esquema 1.
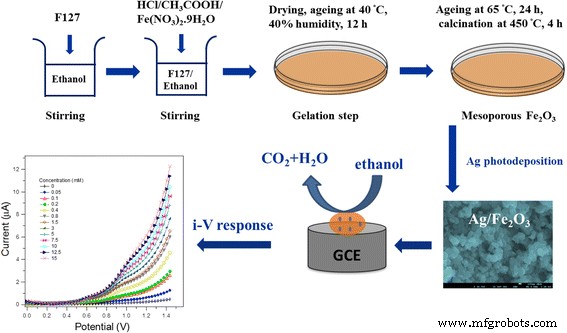
Ilustración esquemática de la síntesis de Ag / α – Fe 2 O 3 -GCE modificado, junto con la detección electroquímica de etanol
Resultados y discusión
Investigación estructural, óptica y morfológica de Mesoporosa Ag / α – Fe 2 O 3
La fase y cristalinidad de los materiales sintetizados fueron examinados en primer lugar por XRD. Como puede verse en la Fig. 1, el espectro XRD del α-Fe 2 derivado de sol-gel O 3 es consistente con el patrón estándar de α - Fe 2 puro O 3 . Todos los picos se pueden asignar perfectamente a la fase cristalina de α – Fe 2 O 3 , (JCPDS-01-086-0550). Además, el patrón XRD no muestra picos de difracción relacionados con otras fases β, γ o δ – Fe 2 O 3 . Además, no se asignaron picos de manera significativa al Ag, lo que podría atribuirse al pequeño contenido de Ag en las muestras preparadas. Otra razón puede deberse al proceso completo de dopaje de Ag en la red del anfitrión, es decir, una difusión de iones en el anfitrión o una migración de iones a la superficie. Dado que el radio iónico de Ag (1,15 Å) es notablemente más alto que el del correspondiente Fe 3+ (0,635 Å), por lo tanto, es razonable considerar la migración de partículas de Ag a la superficie de α-Fe 2 O 3 [35].
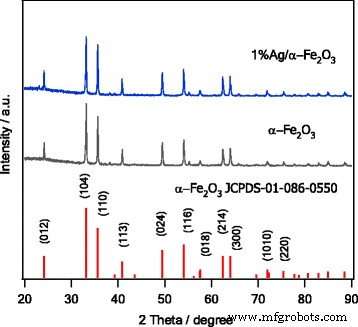
Patrones XRD de α − Fe 2 sintetizado O 3 y 1% Ag / α − Fe 2 O 3 . El patrón estándar de α − Fe 2 puro O 3 también se muestra
La presencia de grupos funcionales adsorbidos en la superficie del α-Fe 2 sintetizado O 3 las partículas pueden examinarse mediante espectroscopía de infrarrojos por transformada de Fourier (FTIR). Como se muestra en la Fig. 2a, la banda observada a ~ 3350 cm - 1 con una pequeña a ~ 1630 cm - 1 se asignan a la vibración de estiramiento de las moléculas de agua, lo que indica la existencia de un poco de agua adsorbida en la muestra. La banda de baja frecuencia a ~ 566 cm - 1 se refiere a la deformación de Fe-O en los sitios octaédricos y tetraédricos de la hematita, lo que da más evidencia de la formación de α-Fe 2 O 3 de acuerdo con los resultados de XRD anteriores. El pico débil a 2900 cm - 1 está relacionado con la banda de estiramiento C – H, lo que significa que algunos compuestos orgánicos no se eliminan por completo de las muestras después de las calcinaciones [40,41,42]. Chen y col. [43] preparó α-Fe hexagonal 2 O 3 nanoestructuras por una fácil reacción alcohol-térmica. Observaron bandas anchas a 3413 cm - 1 y banda débil a ~ 2900 cm - 1 , asignado a las vibraciones de estiramiento de los modos –OH y C – H, respectivamente. Dos picos débiles a 1629 y 1420 cm - 1 correspondiente a la vibración asimétrica y simétrica de los grupos carboxilato, indica una coordinación química del átomo de oxígeno en los aniones acetato con los átomos de hierro en modo unidentado [43]. Además, observaron absorciones fuertes y amplias en el rango de 400 a 700 cm - 1 (440, 530, 570 y 650 cm - 1 ). Estas bandas de absorción se originaron a partir de las vibraciones reticulares inherentes de α-Fe 2 O 3 [43], de acuerdo con el presente trabajo. Por otro lado, Tang et al. [44] demostró un enfoque novedoso hacia el desarrollo de sensores inmunitarios avanzados basados en Fe 3 core-shell funcionalizado químicamente O 4 @Ag nanopartículas magnéticas. Espectro FTIR de Fe 3 puro O 4 mostró los modos vibratorios de estiramiento para el enlace Fe-O a 423 y 572 cm - 1 , mientras que para el Fe 3 recubierto de Ag O 4 el pico a 572 cm - 1 cambiado a 589 cm - 1 y el pico a 423 cm - 1 desapareció por completo, lo que indica la capa de Fe 3 O 4 partículas por Ag.
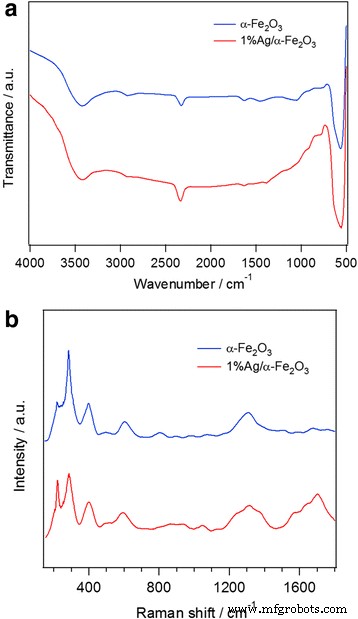
un FTIR y b Espectros Raman de α − Fe 2 O 3 y 1% Ag / α − Fe 2 O 3
Espectros Raman de α – Fe 2 no dopado y dopado con Ag O 3 las muestras se muestran en la Fig. 2b. Los picos espectrales característicos de α-Fe 2 puro O 3 aparecen en 221, 290, 405, 495, 609 y 1315 cm - 1 . Los picos ubicados a 221 y 495 cm - 1 corresponden a la A 1g modo y los picos a 290, 410 y 611 cm - 1 se atribuyen a la E g modo [43, 44, 45]. Generalmente, α – Fe 2 O 3 pertenece al grupo espacial cristalino \ ({D} _ {3d} ^ 6 \) con siete modos de vibración activos Raman, dos A 1g modos (225 y 498 cm - 1 ) y cinco E g modos (247, 293, 299, 412 y 613 cm - 1 ) [45], de acuerdo con el presente trabajo. El pico agudo aparece a ~ 1315 cm - 1 está relacionado con una dispersión de dos magnones que surge de la interacción de dos magnones creados en sitios de espín cercanos antiparalelos [43, 46]. Bhushan y col. [46] observó cuatro picos Raman más a 666, 820, 1050 y 1103 cm - 1 solo en α-Fe 2 altamente cristalizado O 3 en α – Fe 2 alto dopado con Ag O 3 . El presente trabajo exhibe algunos de estos picos, lo que confirma la alta naturaleza cristalina de las muestras preparadas. En la Fig. 2b se observó un pequeño grado de cambio Raman que puede atribuirse a las diferencias tanto en la morfología como en el tamaño de las partículas y / o el estrés. La confirmación de nanopartículas de Ag en caso de 1% Ag / α – Fe 2 O 3 La muestra se evidencia por las bandas ubicadas a 1370 y 1683 cm - 1 [47, 48]. Las intensidades de los picos Raman de α – Fe 2 O 3 es menor que las intensidades relativas de los picos Raman de 1% Ag / α – Fe 2 O 3 lo que puede explicarse por la mejora del campo eléctrico (EF) inducida por la resonancia de plasmón de superficie localizada (SPR) de las nanopartículas de Ag [49]. El efecto electromagnético (EME) asociado con grandes EF locales debido a la excitación de SPR de Ag y un efecto químico (CE) de la interacción electrónica entre Ag y α – Fe 2 O 3 se consideran dos mecanismos de control esencialmente diferentes en el fenómeno de dispersión Raman mejorada en la superficie (SERS). Se entiende que la contribución de EM es varios órdenes de magnitud más que el valor de la mejora química, y la SPR es plasmón de superficie fundamentalmente localizado, en contraste con los plasmones de superficie que se propagan a lo largo de la superficie de Ag. En consecuencia, la SPR de las microestructuras Ag juega un papel principal en el efecto de mejora de SERS [50].
La Figura 3a muestra los espectros UV-vis de α – Fe 2 O 3 y 1% Ag / α – Fe 2 O 3 muestras. En la región ultravioleta (200–400 nm), se observan dos picos de absorbancia alrededor de 270–320 nm. El primero está relacionado con la transmisión electrónica de Fe – O en el mecanismo de contribución de la transición de carga directa de O 2 - 2p → Fe 3+ 3d, y el segundo puede deberse al cambio de forma y tamaño de las partículas [51]. En la región visible (400-800 nm), la absorbancia estrecha a alrededor de 560 nm se origina en la transición de carga indirecta de Fe 3+ 3d → 3d [52, 53]. Además, el cambio en el pico de absorbancia amplio de 424 a 450 nm pico se debe al efecto de resonancia plasmónica superficial de las nanopartículas de Ag, es decir, indica la presencia de nanopartículas de Ag en el α-Fe 2 O 3 [54]. Las intensidades de los picos de absorbancia de α - Fe 2 puro O 3 es superior al 1% Ag / α – Fe 2 O 3 muestra, lo que probablemente se deba a una disminución de la resonancia de Fe-O; la adsorción de oxígeno en las superficies de Ag podría conducir a la formación de óxido de superficie y podría formar especies interactivas Fe-Ag en la muestra híbrida [55]. Zhou y col. [51] estudió las propiedades ópticas del Fe 2 O 3 película fina sintetizada mediante una técnica de sol-gel modificada. Los espectros de transmitancia óptica del Fe 2 O 3 la película mostró un hombro a 500 nm y un pico a 400 nm. El pico del hombro se asigna a la transición de los electrones no enlazantes 3d del Fe 3 + iones a la banda de conducción de acuerdo con el presente trabajo, mientras que el pico se atribuye a la transición de los electrones de enlace 2p del O 2 ̶ iones a la banda de conducción [51].
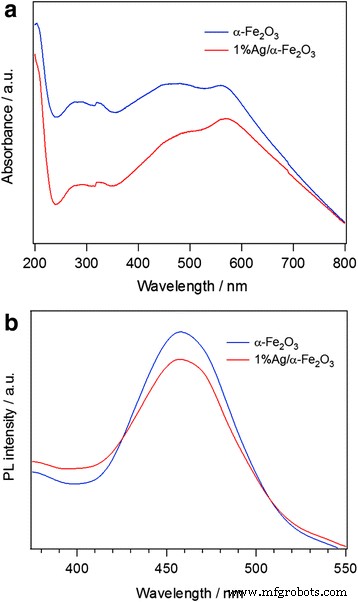
un Espectros de absorción óptica UV-vis medidos en agua DI y b Espectros PL medidos a una longitud de onda de excitación de 315 nm para α − Fe 2 O 3 y 1% Ag / α − Fe 2 O 3
Con el objetivo de investigar los procesos de recombinación de los pares electrón-hueco fotoinducidos, se emplea el análisis espectral de fotoluminiscencia (PL). Los espectros PL de α-Fe 2 puro O 3 y 1% Ag / α – Fe 2 O 3 La estructura híbrida se muestra en la Fig. 3b. Los espectros PL muestran bandas de emisión únicas a una longitud de onda de 460 nm tanto para α – Fe 2 O 3 y 1% Ag / α – Fe 2 O 3 . La intensidad de este pico disminuye notablemente con α-Fe 2 dopado con Ag O 3 muestra de acuerdo con el pico Raman anterior, lo que indica una tasa de recombinación más baja de los pares de electrones y huecos fotogenerados en el Ag / α-Fe 2 O 3 debido a la fuerte capacidad de transferencia de electrones de las nanopartículas de Ag [55,56,57,58]. Kamali y col. [59] observó dos picos PL; el primero ubicado a 710 nm y es amplio e intenso. El segundo es un pico de hombro a 590 nm. Sugirieron que estos picos se deben a la emisión del borde de la banda de α-Fe 2 O 3 nanopartículas [59]. Recientemente, los picos de emisión de PL a 532, 567, 646 y 697 nm observados por Thomas et al. [60]. Estos picos se relacionaron con diferentes bordes de bandas ópticas debido al efecto de confinamiento cuántico.
La Figura 4 muestra la morfología del α-Fe 2 preparado O 3 y 1% Ag / α - Fe 2 O 3 estructura híbrida además del correspondiente análisis químico EDS. Como puede verse, α - Fe 2 puro O 3 La muestra, imagen (a), exhibe una morfología similar a la semiesférica con un tamaño de partícula en el rango de 25 a 70 nm. Además, no se ha logrado ninguna modificación considerable en la forma de las partículas debido a la incorporación de las nanopartículas de Ag; Imagen SEM (b). El patrón espectral EDS confirmó la presencia de nanopartículas de Ag en las nanoestructuras híbridas desarrolladas, con el contenido de carga de Ag que coincidía bien con el experimento.
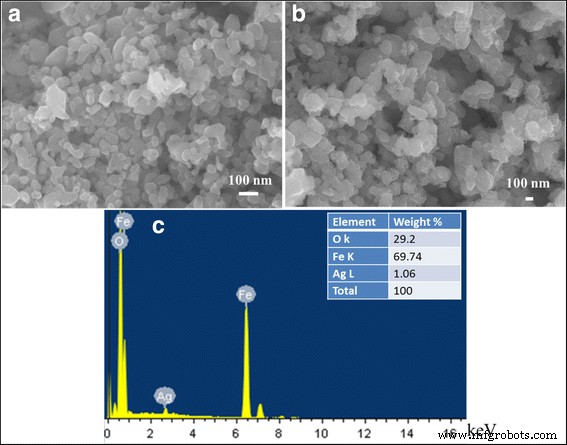
Imágenes SEM de a α − Fe 2 O 3 , b 1% Ag / α − Fe 2 O 3 y c Análisis EDS de 1% Ag / α − Fe 2 O 3 muestra
Se realizó un análisis morfológico detallado utilizando TEM. La Figura 5 presenta la imagen TEM de 1% Ag / α - Fe 2 O 3 y la imagen HR-TEM correspondiente con la difracción de electrones de área seleccionada (SAED). La imagen TEM (a) afirmó el ataque de nanopartículas de Ag a la superficie del huésped Fe 2 O 3 matriz, con tamaños de partícula <20 nm. El α − Fe 2 principal O 3 La matriz reveló nanopartículas esféricas muy finas en el rango de 10 a 30 nm, con algunas esferas más grandes formando una estructura similar a una cáscara y recolectando esas pequeñas nanopartículas en su interior. La imagen HR-TEM (b) de la muestra dopada preparada reveló claramente las franjas de celosía de α − Fe 2 O 3 matriz, junto con la correspondiente a las nanopartículas de Ag. Los espacios interplanares medidos son 0.37 y 0.23 nm correspondientes respectivamente a los planos (012) y (111) de α − Fe 2 hexagonal O 3 celosía y Ag cúbico, confirmando nuevamente la presencia de Ag en la nanoestructura híbrida sintetizada. Como se reveló en el SAED, inserto de la imagen (b), los patrones de difracción muestran diferentes planos de α − Fe 2 cúbica hexagonal O 3 de 012, 104, 113 y 024 correspondientes a d valores de 3,73, 2,70, 2,24 y 1,81 Å, respectivamente.

Imagen TEM de a 1% Ag / α − Fe 2 O 3 y b la imagen HR-TEM correspondiente con el patrón SAED como recuadro
N 2 Se midió la isoterma de adsorción-desorción a 77 K para examinar las propiedades de textura de los materiales sintetizados, como se muestra en la Fig. 6a. Como se reveló, tanto α – Fe 2 O 3 y Ag / α – Fe 2 O 3 mostró un perfil típico de tipo IV con un bucle de histéresis H1, correspondiente a una geometría de poro cilíndrico con una alta uniformidad en el tamaño de los poros y una fácil conectividad de los poros [61]. Un fuerte aumento en el volumen de adsorción de N 2 adsorbido se detectó en P / P0 mayor que 0,8, que está esencialmente asociado con la condensación capilar, lo que indica homogeneidad de la muestra y tamaños de poro pequeños. El área de superficie específica y el volumen de poro total de α – Fe 2 O 3 son 3,55 m 2 / gy 0,004 cm 3 / g, respectivamente, mientras que los valores correspondientes para 1% Ag / α – Fe 2 O 3 son 3,74 m 2 / gy 0,006 cm 3 /gramo. Como puede observarse, se logró un cambio insignificante en las características de textura después de la deposición de Ag. Además, la distribución del tamaño de los poros se muestra en la Fig. 7b. El α – Fe 2 O 3 posee múltiples tamaños de poros con poros dominantes a 8 nm junto con otros poros menores a 4 y 13 nm. El tamaño de poro principal a 8 nm puede estar relacionado con los poros formados inicialmente por la plantilla de copolímero tribloque Pluronic F-127. Se observó una distribución de tamaño de poro bastante similar después de la deposición de Ag, excepto que los poros principales se detectan a ~ 4 nm, probablemente debido a la formación de nanoclusters de Ag.
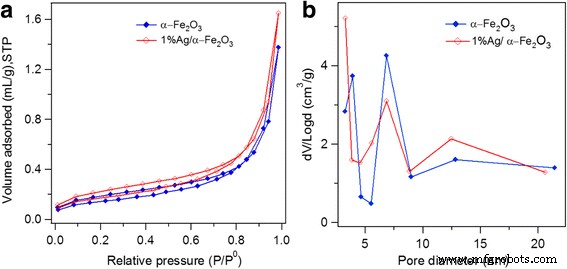
un N 2 isotermas de sorción y b Gráficos de distribución del tamaño de poro de BJH de α − Fe 2 O 3 y 1% Ag / α − Fe 2 O 3
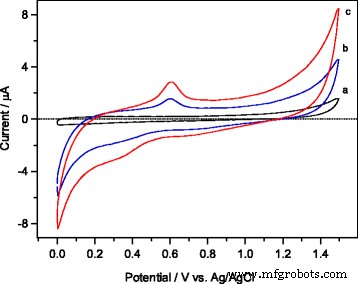
Voltamogramas cíclicos medidos en PBS 0,1 M (pH 7) a una frecuencia de exploración de 50 mVs - 1 en presencia de etanol 5 mM en a GCE desnudo, b Fe mesoporoso 2 O 3 -GCE modificado y c mesoporoso 1% en peso de Ag / Fe 2 O 3 -GCE modificado
Comportamiento electroquímico de electrodos modificados
Para comprender el comportamiento electrocatalítico de los electrodos de trabajo, se aplicó en primer lugar la técnica de voltamperometría cíclica (CV) en una solución tampón de PBS 0,1 M (pH 7) a una velocidad de barrido de 50 mVs - 1 en GCE desnudo, mesoporoso α – Fe 2 O 3 -GCE modificado y mesoporoso 1% en peso de Ag / α – Fe 2 O 3 -GCE modificado usando una concentración fija de etanol 5 mM. Las curvas CV se muestran en la Fig. 7. Como se revela en el gráfico CV de la Fig. 7a, se detectó una pequeña corriente anódica en el caso de utilizar GCE desnudo. Mientras tanto, se observó un aumento significativo en las corrientes anódicas tanto en α-Fe 2 mesoporoso O 3 -GCE modificado (gráfico b) y mesoporoso 1% en peso de Ag / α – Fe 2 O 3 -GCE modificado (gráfico c) en comparación con GCE desnudo (gráfico a), lo que indica una actividad electrocatalítica mejorada de los electrodos modificados. Para comparar ambos electrodos modificados, se observó una corriente anódica máxima de ( I =4.5 μA, gráfico b) para α – Fe 2 puro O 3 GCE modificado, mientras que el 1% en peso de Ag / α – Fe 2 O 3 -Modificado GCE (gráfico c) normalmente genera corriente máxima ( I =8,4 μA), aproximadamente el doble de corriente más que el α-Fe 2 puro O 3 -GCE modificado. Además, durante el escaneo inverso, la corriente catódica probablemente se atribuya a la reducción de agua, y se encontró que esos valores actuales aumentan en el orden de 1% en peso de Ag / α – Fe 2 O 3 > Α – Fe 2 pura O 3 > GCE desnudo. El notable aumento de la corriente anódica sugiere una reacción de transferencia de electrones más rápida y, por lo tanto, permite la detección eficiente de etanol a través de la oxidación al 1% en peso de Ag / α-Fe 2 O 3 -GCE modificado.
Luego se empleó la espectroscopía de impedancia electroquímica (EIS) para investigar las propiedades interfaciales de los electrodos modificados. Gráficos de Bode registrados dentro del rango de frecuencia (0,1 Hz – 100 kHz) en una solución PBS utilizando GCE desnudo, α – Fe 2 O 3 y Ag / α – Fe 2 O 3 Las GCE modificadas se muestran en la Fig. 8. En comparación con α – Fe 2 O 3 o Ag / α – Fe 2 O 3 -GCE modificados, GCE desnudo y sin modificar exhibe una respuesta de impedancia relativamente mayor. Se detectó una reducción en la impedancia en ambos electrodos modificados, lo que indica una actividad electroquímica mejorada. La impedancia más baja con mayor tendencia al proceso de transferencia de electrones se obtiene en el caso de α-Fe 2 dopado con Ag O 3 -electrodo modificado.
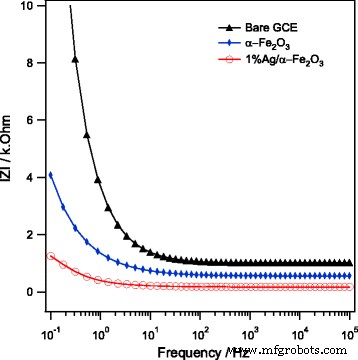
Gráficos de bode de EIS medidos en 0.1 M PBS usando GCE desnudo, α − Fe 2 O 3 y 1% Ag / α − Fe 2 O 3 -GCE modificados a 5 mV de amplitud de potencial, 0,0 V frente a Ag / AgCl en un rango de frecuencia de 0,1 Hz a 100 kHz
Detección electroquímica de etanol en Ag / α – Fe 2 O 3 -GCE modificado
Un potencial de corriente simple ( I-V) La técnica se emplea aquí para examinar y evaluar el comportamiento de detección electroquímica del etanol en los electrodos activos modificados. El I-V respuestas medidas en 1% en peso de Ag / α – Fe 2 O 3 -GCE modificados a 50 mV - 1 en PBS 0,1 M (pH 7) usando diversas concentraciones de etanol (0,05 a 15 mM) se recogen en la Fig. 9a. Como se pudo ver, la corriente anódica aumentó gradualmente al aumentar la concentración de etanol. Este comportamiento electroquímico puede estar relacionado con el aumento de la fuerza iónica de la solución tampón electrolítica de PBS con la concentración de etanol [62]. Más iones en solución podrían proporcionar más electrones a la superficie del electrodo, lo que conduciría a una conductividad mejorada de 1% en peso de Ag / α – Fe 2 O 3 -electrodos modificados [63]. En otras palabras, a concentraciones más altas de etanol, se espera un mayor grado de quimioabsorción de moléculas de etanol, lo que a su vez provocó un cambio considerable en los estados electrónicos en la interfaz electrodo-electrolito y, por lo tanto, se mejora la respuesta de corriente [64]. A partir de los datos de lo anterior ( I-V ) respuesta Fig. 9a, el gráfico de calibración se calculó utilizando los valores de corriente promedio y el resultado obtenido se muestra en la Fig. 9b. Como se reveló, el gráfico de calibración muestra dos pendientes diferentes relacionadas con dos zonas lineales. Tales zonas lineales diferentes corresponden a dos rangos diferentes de concentraciones de etanol:(i) concentración más baja de 0,05 a 0,8 mM y (ii) concentración más alta de etanol de 0,8 a 15 mM. Para una mayor concentración de etanol> 0,8 mM, la corriente anódica presenta un comportamiento lineal con la concentración de etanol pero con una disminución apreciable de la sensibilidad (la pendiente de la zona lineal). La disminución de la sensibilidad observada a concentraciones más altas de etanol probablemente esté relacionada con la saturación de los sitios activos del electrodo con moléculas diana de etanol. Para ambas zonas de concentración, dos ecuaciones lineales ajustadas. (1) y (2) se pueden generar de la siguiente manera:
$$ \ mathrm {en} \ \ mathrm {inferior} \ \ mathrm {concentración} \ \ left ({R} ^ 2 =0.9623 \ right):\ kern3em I \ left (\ upmu \ mathrm {A} \ right) =2.9301 \ \ left [\ mathrm {etanol} \ right] \ \ left (\ upmu \ mathrm {A} \ right) +0.83308 $$ (1) $$ \ mathrm {en} \ \ mathrm {superior} \ \ mathrm {concentración} \ \ left ({R} ^ 2 =0.9876 \ right):\ kern3em I \ left (\ upmu \ mathrm {A} \ right) =0.20793 \ \ left [\ mathrm {etanol} \ right] \ \ left (\ upmu \ mathrm {A} \ right) +3.0807 $$ (2)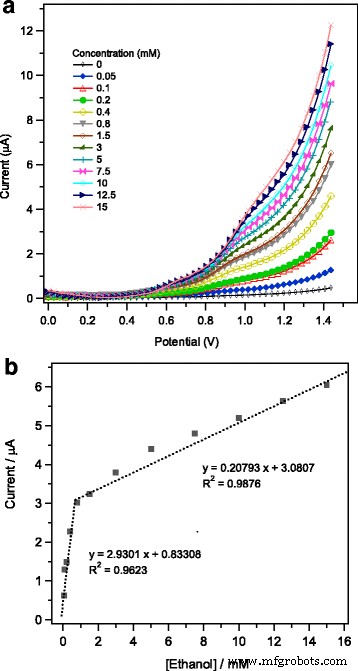
un Típico I-V características de mesoporosa 1% en peso de Ag / Fe 2 O 3 -GCE modificado hacia diversas concentraciones de etanol (de 0,05 a 15 mM), medido en una solución de PBS 0,1 M (pH =7) y b el gráfico de calibración correspondiente
La sensibilidad de Ag / α – Fe 2 O 3 -Se calculó entonces la GCE modificada a partir de la relación de la pendiente de los gráficos de calibración, figura 9b, y el área de la superficie activa del electrodo de trabajo; Se encontró que los valores de sensibilidad eran 41.27 μAmM - 1 cm - 2 en la zona de menor concentración de etanol y 2,93 μAmM - 1 cm - 2 en la zona de mayor concentración de etanol. Vale la pena mencionar que, previamente, se han observado hallazgos de investigación similares de dos regiones de sensibilidad (dos pendientes diferentes) a diferentes concentraciones para la detección de etanol utilizando un electrodo modificado con carbonato de polipropileno / sílice [65] y para el nanocompuesto modificado de Pd / ZnO. CME [66]. Se ha postulado que el fenómeno de dos regiones de sensibilidad se puede explicar de acuerdo con los diferentes modos de adsorción del etanol sobre la superficie del sensor; se produce un proceso de fisisorción en la región de menor concentración que conduce a una mayor sensibilidad del sensor y un modo de quimisorción tiene lugar dentro de la región de mayor concentración que satura la superficie del sensor y, en consecuencia, reduce la sensibilidad [65]. Estas dos zonas lineales diferentes obtenidas con diferentes sensibilidades también se han reconocido durante la detección electroquímica de hidracina en GCE modificado y se discutieron en términos de cambios en el coeficiente de difusión de hidracina debido a la evolución de gas nitrógeno a una concentración más alta de la molécula objetivo [67 ]. En el electrodo modificado por sensor de corriente con Ag / α – Fe 2 O 3 , se observó que al aumentar la concentración de etanol por encima de 15 mM, se logra una saturación de la corriente anódica registrada, lo que finalmente conduce a una región de limitación de detección. El límite de detección (LOD) usando el diseño del sensor de corriente se estimó aplicando la siguiente Eq. (3) [68], teniendo en cuenta la relación señal / ruido de ( S / N =3).
$$ \ mathrm {LOD} =3 {S} _b / m $$ (3)Como se indicó anteriormente en la (Ec. 1), la pendiente del gráfico de calibración en la zona de menor concentración m =2.9301 μAmM - 1 y el valor de ( S b =0.015 μA) es la desviación estándar calculada para una muestra en blanco después de cinco mediciones de corriente. En consecuencia, el LOD se estima en 15,4 μM.
Con el objetivo de examinar la respuesta de detección del electrodo modificado de corriente hacia otros alcoholes, similares I-V Se han realizado experimentos tanto para metanol como para isopropanol en fase líquida. La Tabla 1 recopila las corrientes de oxidación promedio en microamperios, junto con la sensibilidad estimada del electrodo en μAmM - 1 cm - 2 utilizando diferentes soluciones alcohólicas a concentraciones de 0,05, 0,1, 0,2 y 0,8 mM. Como se reveló, el Ag / α – Fe 2 O 3 El electrodo modificado exhibe la respuesta de corriente y la sensibilidad más altas hacia el etanol en comparación con otros dos alcoholes probados. El orden de respuesta del sensor es etanol> metanol> isopropanol.
La cinética de la reacción electroquímica que tiene lugar en la superficie del electrodo durante la detección de etanol se investigó más a fondo mediante la técnica de voltamperometría cíclica mediante la variación de la velocidad de exploración potencial dentro del rango (25-500 mV / s) y midiendo las corrientes anódicas correspondientes. La Figura 10a recopila los voltamogramas cíclicos registrados en el Ag / α – Fe 2 O 3 -GCE modificado en solución de PBS 0,1 M (pH =7) que contiene etanol 0,2 mM a varias velocidades de barrido de 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 450 y 500 mV / s. Como se pudo revelar, se detecta notablemente un aumento gradual en las corrientes pico anódicas con la velocidad de exploración, simultáneamente en la dirección de exploración inversa, las corrientes catódicas aumentan también con la velocidad de exploración. La Figura 10b muestra una buena relación lineal entre las corrientes pico anódicas y la velocidad de exploración, con un coeficiente de correlación ( R 2 =0,9950), lo que indica un proceso cinético controlado por superficie. Además, en la Fig. 10c, los picos de corriente muestran una dependencia lineal de la raíz cuadrada de la velocidad de exploración que da R 2 =0,9954, que es un rasgo característico de una reacción de difusión controlada. Tal estudio de cinética sugiere que la oxidación del etanol en el actual mesoporoso 1% en peso de Ag / α – Fe 2 O 3 La GCE modificada probablemente procede a través de una reacción superficial mixta y una cinética controlada por difusión.
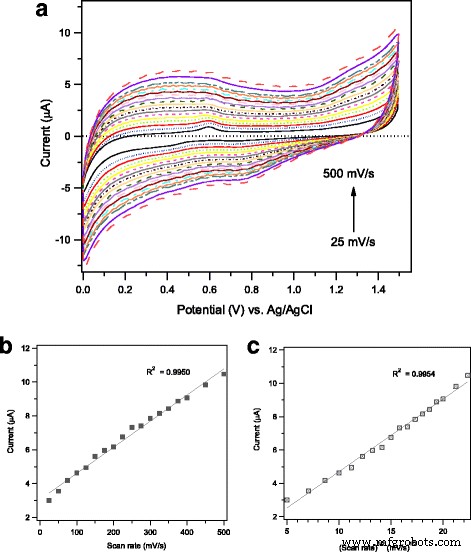
un Voltamogramas cíclicos de mesoporosa 1% en peso de Ag / Fe 2 O 3 -GCE modificado medido en una solución de PBS 0,1 M (pH =7) que contiene etanol 0,2 mM a varias velocidades de barrido de 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350 , 375, 400, 450 y 500 mV / s. Gráfico de la corriente máxima anódica frente a la velocidad de exploración ( b ) y frente a la raíz cuadrada de la velocidad de escaneo ( c )
La Tabla 2 muestra una comparación de los resultados reportados previamente de varios electrodos modificados durante la detección de etanol usando el I-V técnica. La sensibilidad observada aquí usando el electrodo sensor de corriente es significativamente mayor en comparación con las sensibilidades recientemente reportadas, particularmente en el régimen de concentración más baja [62, 65, 66, 69,70,71,72,73,74,75].
Queda una pieza importante de información con respecto a cómo procedería el mecanismo de detección en el actual sistema basado en electrodos modificados. En general, se ha propuesto que las especies de oxígeno quimisorbidas (O - , O 2 - , o O 2 2− ) cubrirá la superficie del electrodo modificado [76]. En consecuencia, se origina una región de carga espacial a través de la extracción de electrones de la superficie del electrodo sensor. Tiene lugar una reacción de superficie entre las especies de oxígeno y las moléculas de etanol adsorbidas, liberando electrones a la banda de conducción de α-Fe 2 O 3 material, Eq. (4) [72] y, por lo tanto, se mejoraron la conductividad y la respuesta del sensor.
$$ {\ mathrm {C}} _ 2 {\ mathrm {H}} _ 5 \ hbox {-} {\ mathrm {O} \ mathrm {H}} _ {\ left (\ mathrm {ads}. \ right) } +6 \ {{\ mathrm {O}} ^ {\ hbox {-}}} _ {\ left (\ mathrm {ads}. \ Right)} =2 {\ mathrm {C} \ mathrm {O} } _2 + 3 {\ mathrm {H}} _ 2 \ mathrm {O} +6 {e} ^ {\ hbox {-}} $$ (4)El Ag metálico y el óxido metálico α – Fe 2 O 3 tendría diferentes sitios activos catalíticos de superficie con comportamiento electroquímico que promoverían los procesos de adsorción y difusión de moléculas de etanol sobre el electrodo de trabajo. Por lo tanto, el rendimiento de detección superior obtenido aquí con el mesoporoso Ag / α-Fe 2 recientemente desarrollado O 3 -La GCE modificada probablemente esté relacionada con la mesoporosidad de α-Fe 2 O 3 , tamaño de partícula pequeño de nanopartículas de Ag con función catalítica, efecto de sensibilización química y electrónica, todo lo cual proporcionaría enormes sitios de adsorción para las moléculas de etanol y promovería el proceso de difusión. A través del dopaje de α – Fe 2 O 3 por nanopartículas Ag, el electrodo modificado basado en sensor de corriente exhibió una sensibilidad extremadamente alta hacia la detección de etanol como 41.27 μAmM - 1 cm - 2 con un LOD muy bajo de 15,4 μM a (S / N =3) a temperatura ambiente.
En aras de la viabilidad del sensor, se evaluó la estabilidad operativa y de almacenamiento junto con la repetibilidad y la reproducibilidad de los electrodos modificados. Usando tres diferentes GCE modificadas activas, los voltamogramas cíclicos registrados en etanol 5 mM dieron una desviación estándar relativa (RSD) ~ 4%, lo que implica una buena reproducibilidad. Cinco pruebas cíclicas sucesivas en la misma solución de etanol arrojaron una RSD <5%, lo que indica una buena repetibilidad del electrodo. Se observó una estabilidad operativa adecuada del electrodo modificado durante su prueba continua durante 45 min en solución de etanol con una reducción menor en la respuesta de corriente. Finalmente, no se requiere ningún cuidado especial para el almacenamiento de electrodos; el presente Ag / α – Fe 2 O 3 GCE modificado mostró una estabilidad de almacenamiento única durante 5 semanas sin casi ningún deterioro de la superficie o reducción de la sensibilidad.
Conclusiones
En resumen, un sensor electroquímico de etanol eficiente basado en Ag / α – Fe 2 mesoporoso O 3 Se han descrito procedimientos sintetizados por un sol-gel fácil y de foto-reducción. El α – Fe 2 mesoporoso O 3 La GCE modificada exhibió una buena actividad electrocatalítica durante la detección de etanol en soluciones tampón de fosfato. Dopaje del material activo α – Fe 2 O 3 por nanopartículas Ag condujo a un rendimiento de detección superior a temperatura ambiente. Una sensibilidad extremadamente alta de 41,27 μAmM - 1 cm - 2 a baja concentración de etanol (0,05 a 0,8 mM) con un LOD muy bajo de 15,4 μM a ( S / N =3) se obtuvo. Además, se encontró que la respuesta de detección y la sensibilidad del electrodo eran mucho más altas para el etanol en compartimentos con metanol o isopropanol. Un rendimiento de detección tan extraordinario probablemente se relacionó con la mesoporosidad de α-Fe 2 O 3 matriz, junto con el pequeño tamaño de partícula de las nanopartículas de Ag. Las características de detección únicas obtenidas en este estudio revelan que el mesoporoso Ag / α-Fe 2 desarrollado actualmente O 3 representaría un material de detección potencial para seguir fabricando sensores electroquímicos de alto rendimiento para la detección de etanol o alcoholes similares en soluciones acuosas.
Abreviaturas
- Ag:
-
Plata
- CV :
-
Voltamperometría cíclica
- EIS:
-
Espectroscopia de impedancia electroquímica
- F-127:
-
Copolímero tribloque plurónico
- GCE:
-
Electrodo de carbono vítreo
- I-V :
-
Actual versus potencial
- LOD:
-
Límite de detección
- m :
-
Pendiente del gráfico de calibración
- PBS:
-
Solución tampón de fosfato
- R 2 :
-
Coeficiente de correlación
- S / N :
-
Relación señal / ruido
- S b :
-
Desviación estándar
- α – Fe 2 O 3 :
-
Hematita (óxido de hierro)
Nanomateriales
- Compuesto híbrido Nanoestructurado de sílice / oro-celulosa-Amino-POSS mediante el proceso Sol-Gel y sus propiedades
- Cómo obtener una cobertura total de la película de perovskita estable mediante el proceso antidisolvente modificado
- Nanofibras poliméricas electrohiladas decoradas con nanopartículas de metales nobles para detección química
- Detección de peróxido de hidrógeno basada en la modificación de superficies internas de nanoporo de estado sólido
- Sustratos de SERS altamente sensibles y de gran superficie con películas delgadas de nanocables de plata recubiertas por un proceso de solución a escala de microlitros
- Sensor plasmónico basado en nanoprismas dieléctricos
- Nanopétalos mesoporosos de óxido de níquel (NiO) para detección de glucosa ultrasensible
- Detección de glucosa electroquímica no enzimática sensible basada en NiO poroso hueco
- Sensor de deformación ultrasensible basado en una película piezoeléctrica de poli (fluoruro de vinilideno) flexible
- Chip de detección química de aproximación al límite cuántico
- Nanosensor químico imprimible