


Efecto del confinamiento en las propiedades fotofísicas de las cadenas P3HT en la matriz de PMMA
Resumen
La influencia de la disposición de las cadenas de poli (3-hexiltiofeno) (P3HT) incrustadas en la matriz de poli (metacrilato de metilo) (PMMA) en las propiedades fotofísicas, como el espectro de absorción electrónico, la banda prohibida y el rendimiento cuántico de fotoluminiscencia, de los agregados de P3HT formados se sido estudiado. Se ha encontrado que la variación de la fracción de P3HT en la matriz de PMMA del 25 al 2% en peso va acompañada del aumento del rendimiento cuántico de fotoluminiscencia, el desplazamiento hacia el rojo de la banda prohibida y el cambio estructural de los cristalitos de P3HT. Los cambios anteriores van acompañados de la ruptura de la red continua de la fracción de P3HT en partículas de P3HT más pequeñas con un tamaño que varía desde varios micrones hasta varias decenas de nanómetros. Los resultados se interpretan en términos del empaquetamiento intermolecular cambiante y la reducción del trastorno torsional intramolecular. Se discute que la mayor contribución a los cambios anteriores proviene de las moléculas de P3HT en la interfaz del grupo de P3HT y el entorno de PMMA.
Antecedentes
La fotofísica de bobinas colapsadas y sistemas confinados a nanoescala de polímeros conjugados ha atraído un interés considerable durante la última década [1, 2, 3, 4]. En particular, los procesos de generación de excitones, recombinación radiativa y transferencia de carga fotogenerada en agregados y cristalitos de nanoescala de poli (3-hexiltiofeno) (P3HT) tienen un impacto directo en el rendimiento de las células solares orgánicas donde este polímero se utiliza como componente activo. Se demostró que la naturaleza de la emisión en moléculas de P3HT aisladas y agregados de P3HT es diferente. La emisión molecular normalmente se origina a partir de un estado de excitón intracadena común correspondiente a la cadena relajada con trastorno de torsión reducido [5]. El espectro de emisión de los agregados de P3HT también se origina a partir de un estado de emisión común, pero correspondiente al excitón singlete intercadena que ha caído por pasos de transferencia de energía únicos o múltiples al dominio con la energía más baja [6]. El rendimiento cuántico (QY) de fotoluminiscencia (PL) de la estructura ordenada de laminillas en películas de P3HT está fuertemente suprimido en comparación con las moléculas libres en soluciones debido a la deslocalización intercadena y la disipación de excitones en el material condensado [7]. Por otro lado, QY puede mejorarse mediante el control de la temperatura [8] o la regioregularidad de las cadenas de P3HT [9]. Se demostró, por ejemplo, que las películas de P3HT regioregular tienen un orden de transiciones ópticas más débiles en comparación con las películas de P3HT regioaleatorio debido a una mayor contribución intercadena para el excitón más bajo en las laminillas en comparación con el carácter intracadena del excitón en P3HT regioaleatorio [9]. Por lo tanto, el desarrollo de estrategias simples y efectivas para manipular las propiedades ópticas de las macromoléculas conjugadas a través de cambios en su diseño intramolecular y ensamblaje y ordenamiento intermolecular tiene un potencial significativo para obtener una mayor comprensión de esta interesante clase de materiales, pero también para su amplia aplicación en la electrónica orgánica.
El objetivo de este trabajo es mostrar cómo la disposición modificada de las cadenas de P3HT influye en las propiedades físicas, como el espectro de absorción electrónico, la banda prohibida y el QY de emisión de las partículas a nanoescala de P3HT. Una estrategia prometedora que permite ajustar las propiedades fotofísicas de las películas de polímero conjugado es la mezcla con el otro polímero inerte. Se sabe que en el caso de P3HT, sus propiedades ópticas pueden verse fácilmente influenciadas por la presencia de un medio huésped adecuado. Por ejemplo, Lee et al. demostraron que las energías de transición óptica en experimentos de absorción y emisión de nanopartículas de P3HT se ven afectadas por un tratamiento hidrotermal (polar) con agua desionizada a temperaturas de hasta 150 ° C en un autoclave [10]. Hellmann y col. mostró que la mezcla de P3HT con el poli (óxido de etileno) (PEO) polar conduce a un aumento de la fuerza del oscilador 0-0, así como a un cambio considerable del espectro de absorción óptica en 0,1 eV [11]. Además, Kim et al. observaron cambios similares en las propiedades ópticas de las nanofibras electrohiladas de P3HT después de mezclar el P3HT y el PEO e hilarlos a partir de mezclas de disolventes polares [12]. Otros estudios han demostrado un pequeño corrimiento al rojo en el espectro de absorción óptica de las películas de P3HT al mezclarlas con poli (etilenglicol) sin la necesidad de aditivos de disolventes polares adicionales [13]. Por tanto, los experimentos anteriores han indicado que las propiedades fotofísicas de P3HT pueden manipularse fácilmente mediante medios de procesamiento. Aunque los estudios anteriores mostraron una influencia significativa del entorno del anfitrión en la banda prohibida de los agregados de P3HT, se ha prestado menos atención a los cambios en la emisión QY. Por ejemplo, Kanemoto et al. han demostrado que el PL de los polímeros conjugados puede mejorarse en estado sólido mediante la dilución utilizando polímeros inertes moderados como el polipropileno [14]. Sin embargo, este efecto se logró mediante la conversión de agregados a la forma molecular del polímero conjugado.
Aquí, demostramos que la mezcla de polímero conjugado P3HT con poli (metacrilato de metilo) polar (PMMA), donde se forman partículas de P3HT de micro y nanoescala, induce cambios sistemáticos en las características físicas de los agregados de P3HT. Mostramos que a medida que disminuye la relación en peso de P3HT a PMMA, la fracción de P3HT demuestra la banda prohibida de desplazamiento al rojo, la mejora en el orden y la mejora en QY de emisión. Mostramos que estos cambios muy probablemente se deben a la planarización de la columna vertebral de los polímeros conjugados en presencia de PMMA bajo la acción de fuerzas hidrofóbicas del material huésped.
Métodos
Preparación de la muestra
Solución madre inicial de P3HT regioregular (~ 93% RR, 99,995% en base a metales traza, con peso molecular promedio en número (M n ) en el rango de 15 a 45 kDa, Sigma-Aldrich) se preparó con una concentración de 1,0% en peso en clorobenceno (CB). Se prepararon mezclas binarias de P3HT y PMMA mediante la adición de una cantidad necesaria de poli (metacrilato de metilo) (PMMA, peso molecular medio (M w ) ) de 120 kDa, Sigma-Aldrich) a solución de P3HT en CB seguido de tratamiento en el baño de ultrasonidos durante 30 min. Las películas se prepararon mediante recubrimiento por rotación sobre sustratos de vidrio a 1500 rpm durante 30 s.
Para los estudios de microscopía electrónica de transmisión (TEM), la película se raspó en el recipiente con acetona, que luego se mantuvo durante varias horas para garantizar que todo el PMMA se disolviera por completo, liberando agregados de P3HT que son prácticamente insolubles en acetona (la solubilidad de P3HT en acetona es menor de 0,1 mg / ml [15]). Se vertió gota a gota una pequeña cantidad de la solución sobre la rejilla de carbón TEM seguido de la evaporación de acetona. La solución de PMMA en acetona se vertió en una cuadrícula separada para obtener imágenes de las muestras de PMMA ordenadas.
Mediciones espectroscópicas
Los espectros de absorción se midieron utilizando espectrofotómetros de doble haz SPECORD M40 y OLIS Cary 14. La placa de vidrio desnudo sirvió como referencia. Los espectros de fluorescencia se recogieron usando un espectrómetro doble SPEX Fluorolog 1680, con una lámpara Xe como fuente de luz. La longitud de onda de excitación se seleccionó a 468 nm. Los espectros de absorción se dan a continuación como normalizados a su máximo para comparar sus características espectrales, y los espectros PL se dan corregidos para la sensibilidad del sistema de registro y normalizados a la absorción de la muestra en la longitud de onda de excitación, es decir, se presentan los espectros PL en términos del QY relativo de la emisión de la muestra.
Las mediciones de la bomba-sonda de absorción transitoria (TA) se realizaron utilizando un sistema de láser Ti:zafiro. La excitación se fijó en una longitud de onda de 410 nm. Las mediciones de TA se llevaron a cabo con la bomba (con una tasa de repetición de 1 kHz y una duración de pulso de ~ 100 fs) y un continuo de luz blanca generado por un cristal de zafiro como sonda. El rayo de la bomba se moduló mecánicamente exactamente a la mitad de la tasa de repetición del sistema CPA (500 Hz), y Δ T / T o ΔOD se detectó con una técnica sensible a la fase utilizando amplificadores de bloqueo. La polarización del haz de la bomba estaba en el ángulo mágico (54,7 °) en relación con el del haz de la sonda. Las señales de transmisión fraccionarias medidas, es decir, TA, vienen dadas por TA =−Δ T / T =( T en - T desactivado ) / T desactivado , donde T en indica la transmisión de la sonda con la bomba encendida y T desactivado la transmisión de la sonda con la bomba apagada. Los espectros obtenidos se rectificaron mediante el procedimiento de calibración de longitud de onda.
Medidas de microscopía
Las morfologías de las muestras se estudiaron tanto por microscopía óptica como por TEM. Se tomaron micrografías ópticas de las muestras utilizando un microscopio óptico BAPU XY-B2 equipado con una cámara fotográfica y una computadora. Los estudios TEM se realizaron utilizando un instrumento JEOL JEM-1400 que funciona a 80 kV.
Resultados
Estudios fotofísicos
Los espectros de absorción electrónica de las películas compuestas de P3HT (Fig. 1a) demuestran un inicio típico de la absorción de aproximadamente ~ 650 nm (1,9 eV) correspondiente a la banda prohibida de los cristales de P3HT, seguido de réplicas vibrónicas a ~ 605, 560 y 525 nm que están relacionados con la transición fundamental (0-0), las bandas laterales (0-1) y (0-2), respectivamente. Hay una evolución gradual en los espectros a medida que disminuye la relación en peso de P3HT a PMMA. Primero, aumenta la relación de amplitud de (0-0) a (0-1) absorción. En segundo lugar, los espectros de absorción muestran un estrechamiento desde el lado de la longitud de onda corta del espectro; esta región se atribuye normalmente a la absorción de moléculas desordenadas en un estado amorfo ya que la absorción molecular de P3HT en soluciones diluidas se observa alrededor de ~ 460 nm; por lo tanto, los cambios anteriores indican una disminución de la fracción amorfa desordenada en la muestra [6, 8]. En tercer lugar, el máximo de absorción relacionado con la transición (0-0) se desplaza gradualmente de 602 a 608 nm; el intervalo de banda, calculado a partir de la intersección de la línea tangente al borde de absorción y el eje de abscisas, también se desplaza al rojo de 1,92 a 1,89 eV a medida que la relación P3HT a PMMA en las películas compuestas disminuye.
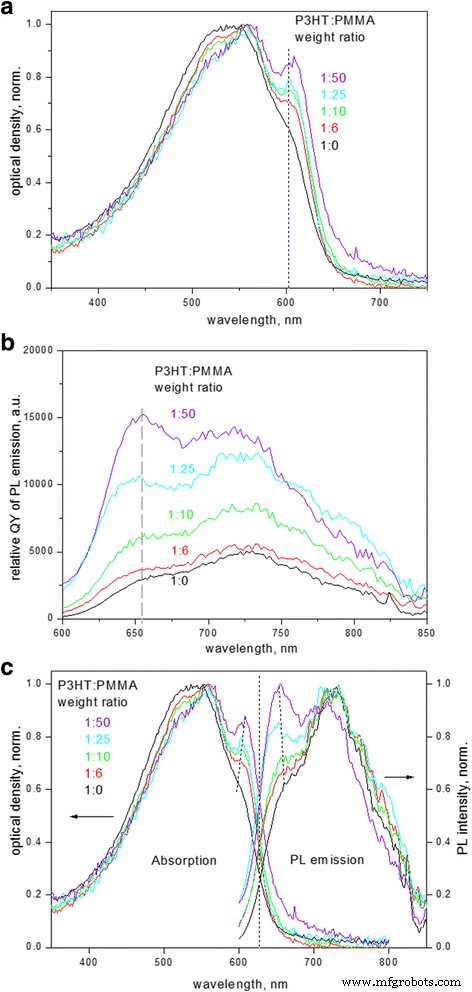
un Espectros de absorción electrónica normalizados, b Espectros PL (en términos de QY relativo) y c comparación de la absorción normalizada y los espectros PL de películas compuestas de P3HT-PMMA con diferentes relaciones de peso de P3HT:PMMA
Los espectros de emisión de PL (Fig. 1b) demuestran un desplazamiento de Stokes de aprox. 0.15 eV, y los espectros tienen un comportamiento similar a los de absorción, con la secuencia espejo de las bandas laterales. La forma de los espectros PL y, en particular, la relación de intensidades de banda (0-0) a (0-1) también dependen de la fracción de P3HT en la matriz de PMMA. Los cambios anteriores se correlacionan bien tanto en la absorción electrónica como en los espectros PL e indican el grado de ordenación en las películas P3HT [16, 17]. La relación de amplitud de (0-0) a (0-1), que es menor que 1, es característica de los agregados H que coexisten con secuencias de cadenas no agregadas [18, 19]. Además, tanto la absorción como los espectros PL de las películas muestran una intensidad creciente del primer máximo con respecto a las bandas laterales (Fig. 1a, b) a medida que disminuye la relación de P3HT a PMMA. El aumento relativo en la intensidad de la transición (0-0) en los espectros evidencia a favor del reordenamiento de las cadenas de P3HT en las películas. La relación de intensidad de las bandas (0-0) a (0-1) está relacionada con el ancho de banda del excitón libre W también, cuya magnitud distinta de cero refleja el grado de desorden en las cadenas de polímero [16] y que se puede calcular usando la Ec. (1) a continuación, asumiendo que un factor de Huang-Rhys es igual a la unidad [20, 6]:
$$ \ frac {A_ {0-0}} {A_ {0-1}} \ approx \ frac {n_ {0-1}} {n_ {0-0}} {\ left (\ frac {1- \ frac {0.24W} {E_p}} {1+ \ frac {0.073W} {E_p}} \ right)} ^ 2 $$ (1)donde n 0 − i es la parte real del índice de refracción en el pico 0 – i y Ep es la energía fonética del oscilador principal acoplada a la transición electrónica. En Eq. (1), la relación del índice de refracción es ∼ 0,97 [6] y la vibración intramolecular principal es E p está dominado por el estiramiento simétrico C =C a 0,18 eV [21]. En cadenas de polímeros más ordenadas, el acoplamiento entre cadenas de Coulombic es más débil, lo que conduce a una reducción del ancho de banda del excitón. El ancho de banda del excitón distinto de cero también afecta la posición de energía del primer máximo de absorción en P3HT, ya que la excitación tiene lugar en el nivel superior de la banda del excitón, mientras que la emisión se produce desde el nivel bajo de la banda, respectivamente. La influencia es la siguiente:cuanto mayor sea el ancho de banda del excitón, mayor será la separación del primer máximo de emisión de PL (asignado como la transición 0-0 en los agregados de P3HT [20]) y el primer máximo de absorción; esta tendencia está indicada por las líneas punteadas en la Fig. 1c.
El ancho de banda de excitón demuestra un estrechamiento a medida que disminuye la relación de P3HT a PMMA (Fig. 2a), acompañado de un aumento de QY de la emisión de P3HT en un factor de cuatro (Fig. 2b). Tal comportamiento debería corresponder, por un lado, al ordenamiento en cadena P3HT. Por otro lado, una disminución en el ancho de banda del excitón está relacionada con el aumento de la correlación intracadena y la disminución de la correlación intercadena [21], lo que implica un aumento simultáneo en el orden intracadena y la longitud de conjugación de la cadena principal del polímero y una disminución en la cantidad de cadenas que participan en π - Interacción π sobre la que se deslocaliza el excitón, acercándose teóricamente a un ancho de banda de excitón cero para una cadena larga idealmente ordenada [22]. Sin embargo, la dependencia del ancho de banda del excitón en la relación PMMA:P3HT en la Fig. 2a puede ajustarse mediante un exponente con un desplazamiento. El desplazamiento indica que el ancho de banda no se acerca a cero; en cambio, llega a un nivel de saturación de 45 ± 5 meV (Fig. 2a). Eso significa que hay un tamaño límite distinto de cero de los agregados de P3HT ordenados en la matriz de P3HT que da lugar a un excitón entre cadenas independientemente de qué tan pequeña sea la fracción de P3HT en la matriz de PMMA. Este hallazgo está relacionado con una fuerte propiedad de los politiofenos regioregulares para autoensamblarse [23] y formar dominios cristalinos altamente ordenados cuyo tamaño característico puede ser tan pequeño como ~ 10 nm [24, 8]. Sin embargo, nuestro valor límite calculado del ancho de banda del excitón es algo mayor en comparación con el observado para los cristalitos de P3HT obtenidos de otros solventes pobres como el mesitileno o el isodureno ( W ~ 30 meV [21]); esta discrepancia puede explicarse en nuestro caso por la presencia de un buen disolvente, es decir, CB, durante la formación de la película del sistema ternario P3HT-PMMA-CB [25].
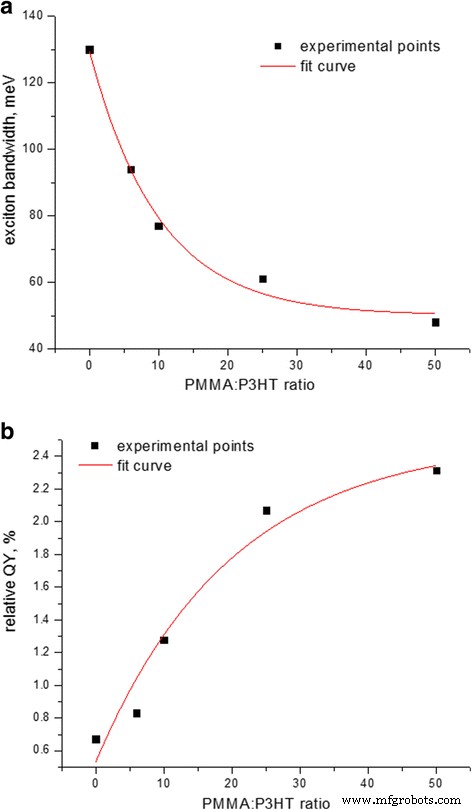
un Ancho de banda de excitación y b QY relativo de emisión de películas compuestas de P3HT-PMMA en función de la relación en peso de PMMA:P3HT, suponiendo que el QY para la película P3HT pura es de aproximadamente 0,5% [25]
La espectroscopía TA proporcionó evidencia adicional sobre el pedido de agregados de P3HT en la matriz de PMMA. Comparación de los espectros TA de las películas compuestas P3HT y P3HT limpias:en la figura 3 se muestran las películas compuestas de PMMA. El espectro típico consta de dos bandas negativas, una de ellas es el blanqueamiento en estado fundamental (GSB) en la región de 530-630 nm, indicativo de blanqueo de absorción 0-1 y 0-0 de P3HT, y la otra banda a ~ 700 nm es indicativa de emisión estimulada (SE). Las bandas positivas en los espectros a ~ 660 y ~ 950 nm son características de la absorción de polarones deslocalizados en dominios cristalinos ordenados y localizados en dominios amorfos desordenados, respectivamente [25, 26, 27]. La banda a ~ 1200 nm se asigna normalmente al excitón singlete TA en P3HT [7, 28, 29, 30]. La diferencia característica en los espectros anteriores (Fig.3) es que las cadenas de P3HT en la matriz de PMMA demuestran la absorción de polarón deslocalizada obvia a ~ 660 nm indicativa de la presencia de regiones ordenadas cristalinas sustanciales de P3HT, mientras que SE pronunciado a ~ 700 nm en la película de P3HT limpia es característica de los excitones intracadena en las cadenas de P3HT desordenadas [8].
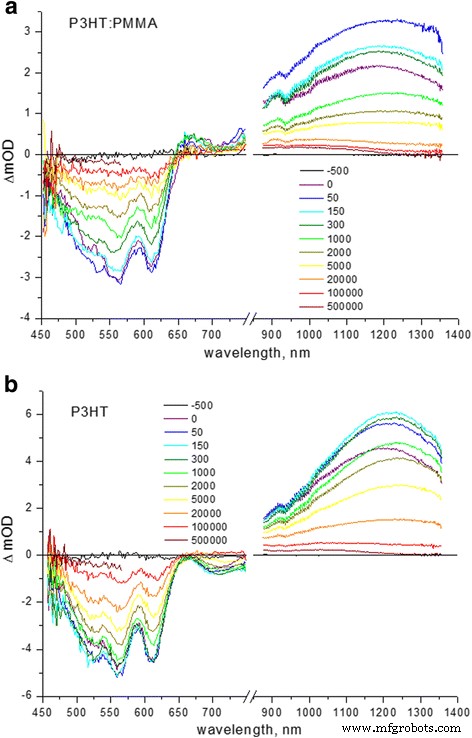
Espectros TA de a P3HT:PMMA (relación de peso 1:50) y b películas pulidas P3HT con recubrimiento giratorio. Los retrasos se indican en femtosegundos en las barras verticales
El análisis de la transición (0-0) y la banda lateral vibrónica (0-1) en los espectros TA revela diferentes dinámicas relativas de relajación en las películas P3HT ordenadas y P3HT-PMMA compuestas (Tabla 1). En la película P3HT ordenada, la banda lateral vibrónica (0-1) decae más lentamente en comparación con la relajación de la transición (0-0), lo que indica la mayor vida útil (con una contribución de ~ 60%) de 7.0 ps para (0-1) transición versus 5.3 ps para la transición (0-0), respectivamente. En el compuesto P3HT-PMMA, la vida útil principal (~ 73%) es más corta y similar para las transiciones (0-1) y (0-0), siendo ~ 1.8 ps, mientras que la vida útil del componente menor (~ 27%) es más lento para las transiciones (0-0) (aproximadamente 300 vs 200 ps, respectivamente), proporcionando una relajación más rápida de la banda lateral vibrónica (0-1) en general (Fig. 3). El componente rápido de relajación del orden de picosegundos es característico de la relajación torsional que conduce a la planarización de la cadena P3HT tras la fotoexcitación [31, 32], mientras que el componente lento es característico de la vida útil de los excitones no fluorescentes que se prueban mediante mediciones de TA [7]. . El diferente comportamiento de relajación en la región GSB para las películas compuestas de P3HT y P3HT-PMMA limpias apunta a favor de una planarización más rápida de las cadenas de P3HT en la muestra compuesta tras la fotoexcitación; eso implica que las cadenas ya se han aplanado parcialmente en el estado fundamental, es decir, tienen menos desorden de torsión en la película compuesta que en la película P3HT pura. Por otro lado, el excitón TA también demuestra una desintegración más rápida en el compuesto P3HT-PMMA, con tiempos típicos de 0,6 ps (68%) y 19 ps (32%) frente a 2 ps (51%) y 40 ps (49%). ) para la película P3HT pura, respectivamente (Tabla 1). El componente ultrarrápido de desintegración anterior puede asignarse a la transferencia de energía del excitón intracadena desde el sitio de alta energía al de baja energía [33] y el componente más lento a la transferencia de energía isoenergética después de que se ha producido la migración rápida del excitón [34]. Parece razonable sugerir que la migración del excitón intracadena avanza más rápido en una cadena más ordenada, libre de desorden torsional, lo que nuevamente confirma que las cadenas P3HT tienen un mejor orden en la muestra compuesta.
Estudios de morfología
Los estudios de microscopía nos permitieron observar la distribución de tamaño de la fracción P3HT en la matriz de PMMA a medida que cambia la relación P3HT:PMMA. En primer lugar, las películas compuestas de P3HT:PMMA revelan una morfología muy estructurada, lo que indica que se produce la separación de fases de P3HT y PMMA, en contraste con la película de P3HT ordenada cuya morfología es relativamente suave (Fig. 4). Sin embargo, a una concentración relativamente alta de P3HT (~ 10% en peso y más), la fracción de P3HT es continua y forma una red de percolación en la matriz de PMMA. A una pequeña concentración de P3HT, la fracción de polímero se transforma para separar partículas de P3HT de tamaño micrónico y submicrónico. La imagen TEM (Fig. 5a) muestra que las partículas pueden ser tan pequeñas como ~ 30 nm y tener prácticamente una forma esférica ideal. La forma esférica de las partículas nos permite sugerir fuerzas repulsivas entre P3HT y PMMA, donde la fase amorfa ("similar a un líquido") de P3HT tiende a separarse de la matriz de PMMA en partículas compactas que poseen una superficie mínima. Por tanto, debería sugerirse una fracción suficiente de la fase amorfa en las partículas de P3HT. El patrón de difracción de electrones de área seleccionada (SAED) (Fig. 5a, a la derecha) muestra la superposición de características amorfas y cristalinas de las partículas. También se han encontrado en la muestra dominios no esféricos altamente cristalinos separados (Fig. 5b). Por lo tanto, se sugiere que la partícula esférica de P3HT consta de un núcleo cristalino rodeado por una fase amorfa de P3HT. Rahimi y col. encontraron que incluso los monocristales altamente ordenados de P3HT están rodeados por una fracción de aproximadamente 12% de moléculas que adoptan una conformación similar a una solución y que el espesor característico de la capa amorfa es de aproximadamente 10 nm [35]. Suponiendo que la capa amorfa de un grosor similar se forma alrededor de un núcleo cristalino, es fácil de entender que una partícula que tiene un tamaño de ~ 30 nm puede adoptar fácilmente una forma esférica debido a una cáscara tan amorfa.
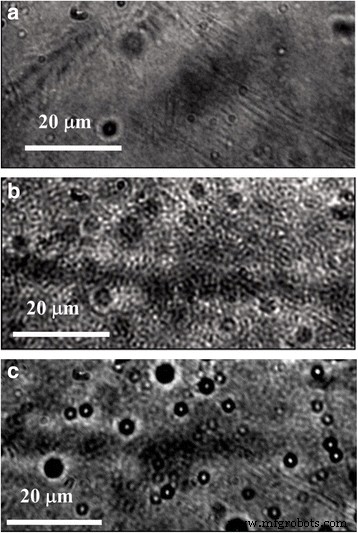
Micrografías ópticas de a P3HT ordenado y b , c P3HT:películas de PMMA con b 10% en peso y c 2% en peso de P3HT en PMMA
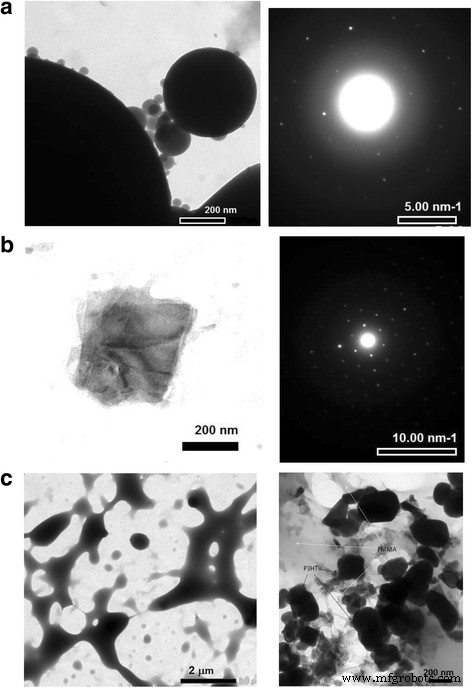
Imágenes TEM de a partículas esféricas de P3HT, b dominio cristalino de P3HT, y sus correspondientes patrones SAED a la derecha de las imágenes; c PMMA ordenado ( imagen derecha ) y mezcla P3HT-PMMA ( imagen izquierda ) se dan a modo de comparación
Asignación de la estructura cristalina
En general, se asume que la textura termodinámicamente favorecida está formada por cadenas de P3HT orientadas de borde a lado [36, 37]. Esta estructura se obtiene en condiciones cercanas al equilibrio realizado para los métodos de formación de película lenta, como la fundición por gota [38, 39], el recubrimiento por inmersión y el recubrimiento por rotación a partir de disolventes de alto punto de ebullición [40]. En muestras preparadas en rejillas TEM, se prefiere la orientación de borde debido a las superficies de rejilla hidrófobas (carbono) que prefieren interacciones con sustituyentes hidrocarbonados de moléculas P3HT. Esta orientación proporciona una "visualización" de principalmente planos (010) y (001) de cristales lamelares P3HT en patrones SAED (Fig. 6).
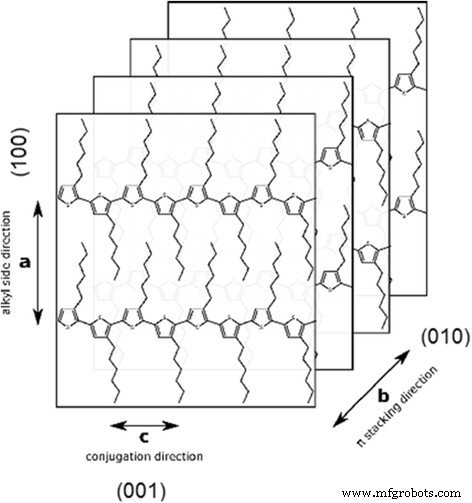
Esquema de la estructura cristalina de P3HT
Los siguientes períodos de apilamiento de cadenas principales sucesivas de politiofeno a lo largo del b Se obtuvieron ejes:0,45 ± 0,1 nm para las estructuras en forma de bola (Fig. 5a, Fig. 7) y 0,48 ± 0,1 nm para la laminilla separada (Fig. 5b). Los valores obtenidos son demasiado grandes para asignarlos a la forma I del cristal P3HT teniendo en cuenta que el ángulo entre a y b los ejes suele estar cerca de los 90 ° [41]. Además, algunos patrones SAED nos permitieron encontrar (h00) anillos de difracción (Fig.8), desde donde es posible determinar la distancia entre apilamientos (la distancia entre cadenas P3HT separadas por capas de n -cadenas laterales de hexilo, es decir, a lo largo de a eje de la celda unitaria monoclínica, Fig.6), siendo de 1,23 nm. La distancia obtenida es característica de la forma cristalina II. Así, si intentamos atribuir los cristales a la forma II, debemos tener en cuenta que el cristal de la forma II tiene un ángulo de inclinación γ =68 ° entre las b eje y los planos del tiofeno [42], desde donde se pueden calcular las distancias interplanar cortas en 0,417 y 0,445 nm, respectivamente. El último valor concuerda bien con la distancia interplanar corta en la forma cristalina II (0.44 nm [43]), mientras que el primero corresponde mejor a una forma intermedia I 'con la distancia interplanar de 0.41-0.42 nm [44].

un Imagen TEM y b SAED de un dominio P3HT con forma de bola en una muestra de mezcla de P3HT-PMMA (relación de peso 1:50)

SAED de un dominio P3HT en una muestra de mezcla P3HT-PMMA (relación de peso 1:50)
Discusión
El principal hallazgo de este trabajo es que el QY de emisión de la fase condensada de P3HT puede mejorarse no debido al desenredo de agregados apretados con una extinción sustancial de excitones por moléculas vecinas en la forma molecular de P3HT, sino por simple reducción del tamaño de P3HT condensado. fase a micro y nanopartículas. Se pueden considerar dos razones principales que son responsables del fenómeno anterior:Primero, el aumento en el área de interfaz de P3HT / PMMA, donde las moléculas interfaciales aumentan su contribución a las propiedades de emisión debido al aumento de la relación superficie-volumen en la disminución. Partículas de P3HT; En segundo lugar, la disposición modificada de las cadenas de P3HT en los dominios cristalinos como resultado de las fuerzas repulsivas que actúan desde el PMMA, que afectan a más moléculas de P3HT a medida que disminuye la proporción de P3HT a PMMA.
La primera razón posible implica el cambio en la constante dieléctrica del entorno P3HT. De hecho, Hu et al. informaron que el reemplazo de un solvente relativamente polar con una constante dieléctrica alta (ε> 3) por uno no polar con una constante dieléctrica baja (ε <3) conduce a una mejora en la fluorescencia QY de los agregados de P3HT en casi un orden de magnitud [45]. Cabe señalar que el PMMA tiene ε> 2,8 [46] y, en principio, se puede considerar que afecta la QY de la emisión de PL. Para verificar la contribución de este factor, verificamos la emisión de PL de P3HT a medida que el ambiente del solvente fue reemplazado gradualmente por moléculas de PMMA (Fig. 9). En el primer experimento, se añadieron las mismas cantidades de la solución madre de P3HT a las cubetas con CB y solución concentrada de PMMA en CB, respectivamente, donde los volúmenes de solución eran los mismos (Fig. 9a). En el segundo experimento, para eliminar un posible error casual del volumen de la jeringa que suministra una solución de P3HT, se añadió polvo de PMMA a la solución de P3HT y se midieron los espectros sucesivamente durante la disolución de PMMA (Fig. 9b). Ambos experimentos mostraron un aumento pequeño pero distinto en el QY relativo de emisión de la solución de P3HT en presencia de PMMA. Por lo tanto, los cambios en la constante dieléctrica de CB (ε ~ 5,6) a la de la mezcla CB-PMMA y luego al ambiente limpio de PMMA en películas delgadas deberían facilitar una mayor fluorescencia QY. Sin embargo, se evaluó que este efecto en las soluciones era pequeño, lo que inducía un aumento en QY de PL solo en ~ 14%. Por otro lado, en las películas, se encontró que el aumento en QY de PL era de hasta ~ 400% (Fig. 2b). Por lo tanto, el cambio relativo en la constante dieléctrica solo tiene un efecto débil concomitante sobre la mejora de QY de PL en películas compuestas delgadas.
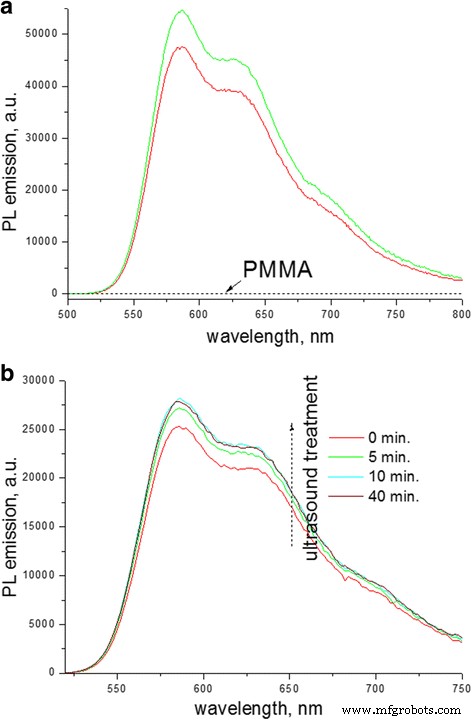
Espectros de emisión PL (λ exc =468 nm) de solución de P3HT (0.01% en peso) en CB: a antes ( rojo ) y después ( verde ) mezclar con una solución de PMMA (5,4% en peso), también se proporciona el espectro PL de PMMA; b tras la adición de polvo de PMMA a la solución pura de P3HT ( rojo ) seguido de un tratamiento sucesivo de la cubeta en un baño de ultrasonidos y una disolución gradual de PMMA hasta ~ 3,25% en peso
El otro factor importante que se puede inferir particularmente de los cambios espectrales de las películas de P3HT-PMMA es el cambio en la disposición mutua de las cadenas de polímero en los dominios P3HT que se encuentran en la matriz de PMMA. Cabe señalar que los cristales de P3HT pueden adoptar diferentes formas, es decir, la forma I que se observa con mayor frecuencia en películas delgadas después del recocido [47], o la forma II, que representa una situación energéticamente más estable [42]. La Forma II puede obtenerse, por ejemplo, mediante la acción sinérgica de la matriz de polímero hidrófilo y un disolvente pobre como el agua en las cadenas de P3HT durante la formación de la película [11], y muestra un notable desplazamiento hacia el rojo en el espectro de absorción [35]; Se observa una tendencia similar en nuestros resultados, que muestra un desplazamiento hacia el rojo de la banda prohibida de 1,92 a 1,89 eV (Fig. 1a). Curiosamente, la distancia de apilamiento π - π informada para los cristales nanofibrilares P3HT de la forma II es relativamente grande, de 0,440 nm [43], en comparación con la distancia de apilamiento encontrada para la forma I que está entre 0,340 y 0,414 nm [48,49 , 50]. Al mismo tiempo, hay una interdigitación de la cadena lateral de alquilo más estricta en la forma II, con la distancia entre cadenas (en la dirección en la que apuntan los grupos alquilo) de 1,20 a 1,31 nm [42] frente a la que varía de aproximadamente 1,55 a 1,73 nm en cristales de forma I [48, 50]; la interdigitación más estricta parece estabilizar mejor el orden intracadena en cristales de forma II.
La discusión anterior sobre las diferentes formas cristalinas de P3HT es importante para comprender la transformación estructural de los cristales de P3HT formados en la matriz de PMMA a diferentes relaciones en peso de P3HT a PMMA. Primero, se ha encontrado que la posición máxima relacionada con la banda (0-0) en películas de P3HT-PMMA con recubrimiento giratorio experimenta un ligero desplazamiento hacia el rojo en una pequeña proporción de P3HT a PMMA, es decir, de 602 a 608 nm (Fig. 1a) . En segundo lugar, los estudios de microscopía nos permitieron distinguir dos tipos de cristales en las muestras de mezcla, que tienen distancias interplanar cortas en la dirección de apilamiento (a lo largo de la b eje del cristal P3HT) sea de 0,417 (que es característico de las estructuras en forma de bola, ver Fig. 5a y Fig. 7) y 0,445 nm (característico de la estructura de laminillas mostrada en la Fig. 5b), respectivamente. The latter value agrees well with the crystalline form II as discussed above, whereas the former one better corresponds to an intermediate form I’ reported by Roehling et al. [44], which possesses the interplanar distance of 0.41–0.42 nm. They also showed that the form I’ demonstrates an increase in the coherent domain size in the π − π stacking direction by a factor of ~ 2 (from 6.3 to 12.4 nm), as compared to form I in samples prepared from p-xylene, which can be responsible for the enhancement of the (0-0) band relatively to the (0-1) band in P3HT samples [50].
Based on the above discussion, we can conclude that the composite P3HT-PMMA samples contain crystals of both forms of P3HT (I’ and II) because the interchain stacking distance varies from 0.42 to 0.44 nm for crystals of the different morphology. Thus, it can be suggested that the changing (0-0) to (0-1) ratio is related to the changing weight ratio of the different crystalline forms of P3HT, respectively, and the increasing (0-0) to (0-1) ratio most probably is due to increasing fraction of the form I’ in the blend, which promotes the increasing coherent domain size in the π − π stacking direction of P3HT domains. The reason of the above variations is tentatively assigned to hydrophobic forces acting on P3HT chains being in the polar environment, i.e., PMMA matrix, which forces P3HT aggregates to conform a specific arrangement inside the matrix. Such a process is most effective for smaller P3HT particles since the most influence is rendered onto the molecules being at the interface of P3HT-PMMA. Additional evidence that supports the above suggestion is the fact that the ratio of the first absorption maximum in respect to the sidebands decreases with time, which implies that the equilibrium between the different crystalline forms of P3HT in PMMA matrix evolves, namely, the form I’ gradually converts to the more thermodynamically stable form II (Fig. 10). Such a result reflects slow relaxation processes in PMMA matrix itself that acts on P3HT domains and thus renders a delayed effect which is more pronounced in samples with smaller P3HT particles.

Electronic absorption spectra of as-prepared samples (solid curves ) and the same samples 2 weeks later (dotted curves ) of P3HT-PMMA (1:50 weight ratio, red lines ) and P3HT-PMMA (1:4 weight ratio, blue lines )
Conclusions
An increasing QY of PL which has been found in P3HT particles embedded into PMMA matrix is an unusual phenomenon since it takes place when the polymer molecules are still aggregated and where a strong exciton quenching should be normally observed. The increasing QY is assigned due to the two factors. The minor factor is the changing dielectric constant which facilitates a modest increase in QY by about 14%. The major factor is due to rearrangement of the polymer chains themselves. Better chain ordering in P3HT domains embedded into the PMMA matrix has been unequivocally proved by spectroscopy methods and calculation of the exciton bandwidth as well. The reason of the structural changes is tentatively assigned to hydrophobic forces acting on P3HT chains being in polar environment, i.e., PMMA matrix, which forces P3HT aggregates to conform a specific arrangement inside the matrix. Such a process is most effective for smaller P3HT particles since the most influence is rendered onto the molecules being at the interface of P3HT-PMMA. Particularly, it can be concluded that the composite P3HT-PMMA samples contain P3HT crystals of two forms, i.e., I’ and II, in which the interchain stacking distance varies from 0.42 to 0.44 nm. In form I’, intramolecular torsional disorder is reduced and most probably it promotes the increasing coherent domain size in the π − π stacking direction of P3HT domains, respectively. This is accompanied by the increasing first absorption maximum in respect to sidebands in spectra of composite P3HT films and by narrowing free exciton bandwidth, respectively. It is interesting to note that the narrowing exciton bandwidth is a factor which is responsible for increasing QY of PL emission in semiconductor nanoparticles as compared to the bulk crystals possessing wide energetic bands [51]. Narrow bands reduce smearing effect upon electronic transitions, thus facilitating more electrons falling to the same energy level. Thus, the observed enhanced QY of emission of P3HT particles can be interpreted in terms of the changing intermolecular packing and reduced intramolecular torsional disorder along with narrowing exciton bandwidth.
Nanomateriales
- Propiedades aritméticas
- Nanopartículas semiconductoras
- El efecto del plasma de no equilibrio por contacto sobre las propiedades estructurales y magnéticas de Mn Х Fe3 - X О4 Espinelas
- Efecto del polietilenglicol en el fotocátodo de NiO
- Propiedades paramagnéticas de nanomateriales derivados del fullereno y sus compuestos poliméricos:efecto de bombeo drástico
- Efecto sinérgico del grafeno y los MWCNT en la microestructura y las propiedades mecánicas de los nanocompuestos de Cu / Ti3SiC2 / C
- Nanocompuestos magnéticos de poli (N-isopropilacrilamida):efecto del método de preparación sobre las propiedades antibacterianas
- Efecto del método de síntesis de nanopartículas de manganita La1 - xSr x MnO3 en sus propiedades
- MATLAB - Matriz
- C# - Propiedades
- Propiedades de la madera