


Dependencia de la composición de las propiedades estructurales y electrónicas de InGaNBi cuaternario
Resumen
Para realizar una ingeniería de estructura de banda factible y, por lo tanto, una eficiencia de luminiscencia mejorada, InGaNBi es una aleación atractiva que puede explotarse en dispositivos fotónicos de luz visible e infrarrojo medio. En el presente estudio, las propiedades estructurales y electrónicas como la banda prohibida, la energía de división de la órbita de espín y la deformación del sustrato de las composiciones de InGaNBi frente a In y Bi se estudian mediante el uso de cálculos de primeros principios. Los parámetros de la red aumentan casi linealmente con el aumento de las composiciones de In y Bi. Mediante el dopaje con bismuto, la banda prohibida cuaternaria de InGaNBi podría cubrir un amplio rango de energía de 3.273 a 0.651 eV para Bi hasta 9.375% e In hasta 50%, correspondiente al rango de longitud de onda de 0.38-1.9 µm. La energía de división de espín-órbita calculada es de aproximadamente 0.220 eV para 3.125%, 0.360 eV para 6.25% y 0.600 eV para 9.375% Bi, respectivamente. También hemos mostrado la cepa de InGaNBi sobre GaN; indica que mediante el ajuste de las composiciones de In y Bi, InGaNBi se puede diseñar en GaN con una cepa aceptable.
Introducción
En los últimos años, la wurtzita (WZ) En x Ga 1− x Las aleaciones de N y los pozos cuánticos (QW) de InGaN / GaN han despertado una gran atención debido a su gran potencial para desarrollar células solares, diodos emisores de luz (LED) de alta eficiencia y diodos láser (LD) [1-10]. El In de uso común orientado a [0001] x Ga 1− x Los QW de N / GaN sufren un intenso campo eléctrico incorporado inducido por la tensión de compresión biaxial del In x Ga 1− x Capa N [11], que da lugar a la disminución de la energía de emisión QW y la fuerza del oscilador de los pares electrón-hueco. Además, existe una alta densidad de defectos geométricos en En x Ga 1− x N aleaciones, incluidas las fallas de apilamiento y las dislocaciones de subprocesamiento (TD) [12]; estos TD tienen una gran correlación con los centros de recombinación no radiativa. Los defectos, la fuga de electrones y la recombinación Auger son las tres fuentes de la caída de eficiencia de In x Ga 1− x N LED, de los cuales la recombinación Auger es la causa principal [13].
De manera similar, para los diodos infrarrojos basados en GaAs, ya se ha propuesto que la aleación de bismuto es un método eficaz para disminuir la banda prohibida ( E g ) así como mejorar la división de espín-órbita (SO) para lograr la supresión del proceso de recombinación de Auger [14]. El elemento más grande del grupo V del bismuto revela efectos atractivos sobre las propiedades físicas de las aleaciones de bismuro. Los cambios en la estructura de la banda de las aleaciones de bismido se han investigado para diferentes materiales de aleaciones ternarias tanto de manera experimental como teórica, como AlNBi [15], GaNBi [16, 17], GaSbBi [18, 19], InPBi [20, 21], e InSbBi [19, 22-24]. La banda prohibida se modifica principalmente por la gran cepa inducida por átomos de Bi a alta concentración en InPBi. La incorporación de Bi perturba las bandas de valencia (VB) debido a la interacción de los estados de impureza de Bi con las bandas de agujeros pesados / ligeros y las bandas de separación de la órbita de espín [21]. Más recientemente, las aleaciones de bismido cuaternario (por ejemplo, GaAsNBi [25-27], InGaAsBi [28, 29], GaAsPBi [30]) también han atraído una gran atención. Las distorsiones locales alrededor de los átomos de P y Bi contribuyen significativamente a la modificación de banda prohibida de GaAsPBi. Un requisito de composición para Ga As 1− x - y P y Bi x para lograr una relación de recombinación Auger más baja que GaAs se obtuvo [30]. La combinación de bismuto y otros átomos III o V aumenta el alcance de la ingeniería de la estructura de la banda, incluido el control de la banda prohibida, la división de la órbita del espín, la conducción (CB) y las compensaciones de la banda de valencia y la deformación [25]. Por lo tanto, es de gran interés describir el efecto de la sustitución de Bi en el [0001] In x Ga 1− x N / GaN, sintonizando las propiedades estructurales y electrónicas y, por tanto, la eficiencia de la luminiscencia. En el presente estudio, utilizando cálculos de primeros principios [31], las propiedades estructurales y electrónicas como la banda prohibida, la energía de división de espín-órbita ( Δ SO ), y se estudian la cepa de sustrato de InGaNBi frente a las composiciones de In y Bi. Teniendo en cuenta el gran desajuste de la red y la mala calidad del contenido de In superior al 55-60% en la muestra de InGaN [32], así como la baja solubilidad del bismuto en las aleaciones de bismuro diluido, las concentraciones de In y Bi se controlan hasta en un 50% y 9,375%, respectivamente. El documento está organizado de la siguiente manera. En la sección "Métodos", presentamos los métodos computacionales detallados. Las propiedades estructurales, electrónicas y la tensión del sustrato se proporcionan en la sección "Resultados y discusión". Finalmente, se resume un breve resumen.
Métodos
Nuestros cálculos teóricos se basan en la teoría funcional de la densidad (DFT) [31] implementada en el código VASP [33, 34]. En los cálculos de propiedades estructurales, las interacciones electrón-ión y de intercambio-correlación se tratan con el método de onda aumentada del proyector (PAW) [35, 36] y la aproximación de gradiente generalizada (GGA) de Perdew-Burke-Ernzerhof (PBE). [37], respectivamente. Las configuraciones de electrones de valencia para átomos de In, Ga, N y Bi se emplean como 4 d 10 5 s 2 5 p 1 , 3 d 10 4 s 2 4 p 1 , 2 s 2 2 p 3 y 5 d 10 6 s 2 6 p 3 , respectivamente. Con el fin de superar la subestimación del potencial PBE en la banda prohibida de las propiedades electrónicas, empleamos el potencial de intercambio de Becke-Johnson modificado en combinación con la correlación de aproximación de densidad local (MBJLDA) [38]. El bismuto tiene un gran efecto de acoplamiento de espín-órbita (SOC) y, por lo tanto, el SOC se incluye en los cálculos electrónicos. En todos los cálculos, las estructuras se relajan hasta que las fuerzas en cada átomo se vuelven menos de 0.02 eV / Å y el cambio de energía máximo es del orden de 10 −4 eV. Se establece un corte de onda plana de 450 eV para garantizar la precisión de los cálculos. Un paquete Monkhorst de 4 × 4 × 4 k Se adopta una malla de puntos en la primera zona de Brillouin.
Resultados y discusión
Propiedades estructurales
Las supercélulas constan de 4 × 2 × 2 de células primitivas WZ-GaN, incluidos 64 átomos. Investigamos 36 composiciones de I n y Ga 1− y N 1− x Bi x con 0≤ x ≤0.09375,0≤ años ≤0,5 basado en experimentos recientes en los que la muestra de InGaN presenta un gran desajuste de la red y una calidad deficiente para un contenido de In superior al 55-60% [32], así como la baja solubilidad del bismuto en las aleaciones de bismuro diluido. Se considera una configuración representativa en la que los átomos de In y Bi se distribuyen uniformemente. Hemos resumido los parámetros de celosía calculados de ternario In y Ga 1− y N y cuaternario En y Ga 1− y N 1− x Bi x aleaciones junto con otros datos teóricos y experimentales en la Fig. 1. Para GaN prístino, los parámetros de celosía a =3.211, c =5.235 Å, que concuerdan con otros cálculos teóricos a =3.155,3.22 Å, c =5.144,5.24 Å [39–41] y datos experimentales 3.19 Å para a , 5,19 Å para c [42]. Los parámetros de celosía ( a , c ) de En y Ga 1− y El N aumenta cuando la composición de In aumenta y muestra una variación casi lineal, como se muestra en la Fig. 1a. Los cálculos actuales predicen a =3,304 Å, c =5.365 Å para En 0,25 GaN y a =3.397 Å, c =5.509 Å para En 0.5 GaN, todos los cuales concuerdan bien con los resultados anteriores de a =3,33 Å, c =5,39 Å para En 0,25 GaN y a =3.43,3.485 Å, c =5.55,5.488 Å para I n 0.5 GaN [39, 40, 43, 44]. En el caso de aleaciones cuaternarias In y Ga 1− y N 1− x Bi x En lo que a nosotros respecta, no existen valores experimentales y teóricos para las propiedades estructurales. En la Fig. 1b, los parámetros de la red obtenidos también aumentan casi linealmente con el aumento de las composiciones de In y Bi. Debido a los radios iónicos más grandes de In y Bi que los átomos de Ga y N, la sustitución de In sobre Ga y Bi sobre N conduce a parámetros de red mejorados de InGaNBi.
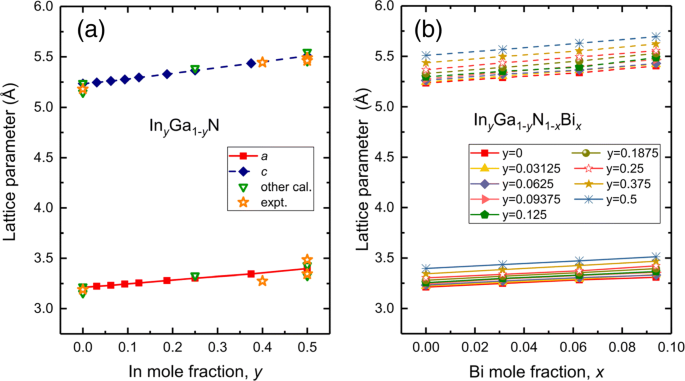
Los parámetros de celosía para a aleaciones ternarias En y Ga 1− y N , con 0≤ y ≤0,5 y b aleaciones cuaternarias En y Ga 1− y N 1− x Bi x , con 0≤ x ≤0.09375, 0≤ años ≤0,5. A modo de comparación, agregamos algunos otros cálculos y datos experimentales de la Ref. [39-44] en la figura 1a. La línea continua representa a y la línea discontinua es c
La incorporación de In y Bi romperá la periodicidad del cristal e introducirá deformaciones geométricas en una estructura fuertemente aleada. Elegimos En 0,25 GaN Bi 0.0625 como ejemplo de cuatro estadísticas de enlaces químicos, como se muestra en la Fig. 2; las longitudes promedio de los enlaces Ga-N, In-N, Ga-Bi e In-Bi son 2.009, 2.195, 2.592 y 2.704 Å, respectivamente. Tenga en cuenta que la longitud del enlace Ga-N en el GaN a granel prístino es 1.970 Å. La longitud del enlace In-N es mayor que la de Ga-N, lo que indica que el átomo de In aleja notablemente el átomo de N. De manera similar, la mayor longitud de enlace de Ga-Bi que Ga-N significa que el átomo de Bi empuja al átomo de Ga, encontrando una buena consistencia con el orden de radios covalentes de Ga (1.22 Å), In (1.42 Å), N (0.71 Å), y Bi (1,48 Å) [45]. Otras configuraciones muestran un comportamiento similar. La deformación de la celosía y la disparidad en la electronegatividad entre el huésped y el dopante tienen un efecto considerable en las propiedades electrónicas y ópticas.
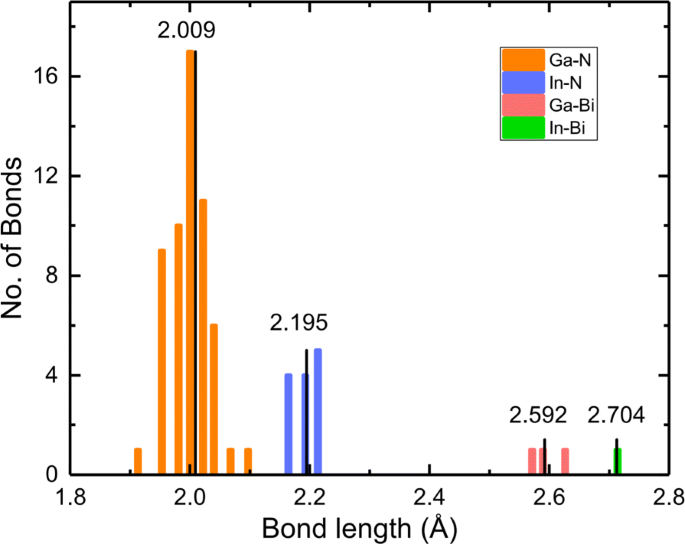
Histograma de la longitud del enlace en In 0,25 GaNBi 0.0625 . Los valores en el panel indican las longitudes promedio de los cuatro tipos de enlace
Propiedades electrónicas
Se ha demostrado que los potenciales funcionales o de corrección y el efecto SOC influyen en gran medida en la precisión prevista de la energía de banda prohibida de la aleación III-V, la banda de valencia y la energía de división de la órbita de espín. Por lo tanto, validamos nuestros resultados utilizando el potencial MBJLDA y los comparamos con otros cálculos y experimentos teóricos. La figura 3 es un gráfico de la energía de la banda prohibida frente a la composición de In en In y Ga 1− y N así como un ajuste a los datos. También se grafican algunos valores de banda prohibida obtenidos mediante experimentos, funciones teóricas de HSE06, mBJ y LMTO-CPA-MBJ. La banda prohibida predicha de GaN es 3,273 eV, que está en buena coherencia con los cálculos y experimentos actuales, 3,33 eV por mBJ [40], 3,261, 3,23 eV por HSE06 [39, 46] y 3,40-3,50 eV por experimentos [47– 49]. Como se observa en I n y G a 1− y N, nuestros resultados de DFT confirman que E g valores de I n y G a 1− y N disminuye continuamente a medida que y aumenta de 0 a 50%. E g disminuye suavemente de 3,273 a 1,546 eV. Esto se compara bien con los resultados teóricos (HSE06, potenciales mBJ) [39, 40, 46] y experimentales [50, 51].
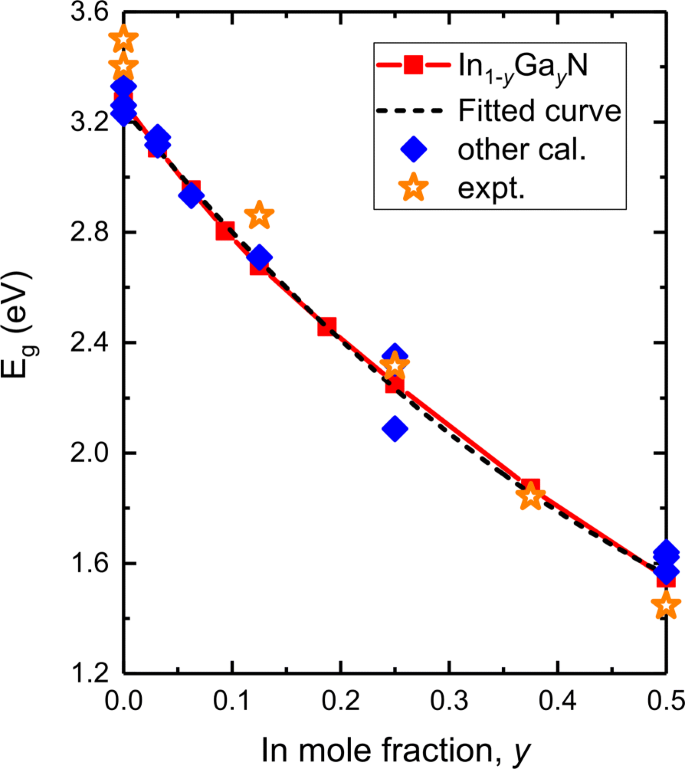
Energía de banda prohibida prevista ( E g , línea sólida roja) en función de En composición en I n y G a 1− y N así como un ajuste a los datos (línea discontinua negra). También se grafican otros resultados teóricos [39, 40, 46] y experimentales [47-51]
La gráfica de contorno para la banda prohibida del cuaternario I n y G a 1− y N 1− x B yo x Las aleaciones se muestran en la Fig. 4. Las bandas prohibidas de las aleaciones cuaternarias muestran una tendencia no lineal en función de la composición, que disminuye al aumentar los contenidos de In y Bi. A partir de los resultados, encontramos que la banda prohibida de InGaNBi podría cubrir un amplio rango de energía de 3.273 a 0.651 eV para Bi hasta 9.375% e In hasta 50%, correspondiente al rango de longitud de onda de 0.38 a 1.9 µm, indicando sus potenciales aplicaciones optoelectrónicas en Alcance de luz visible e infrarrojo medio.
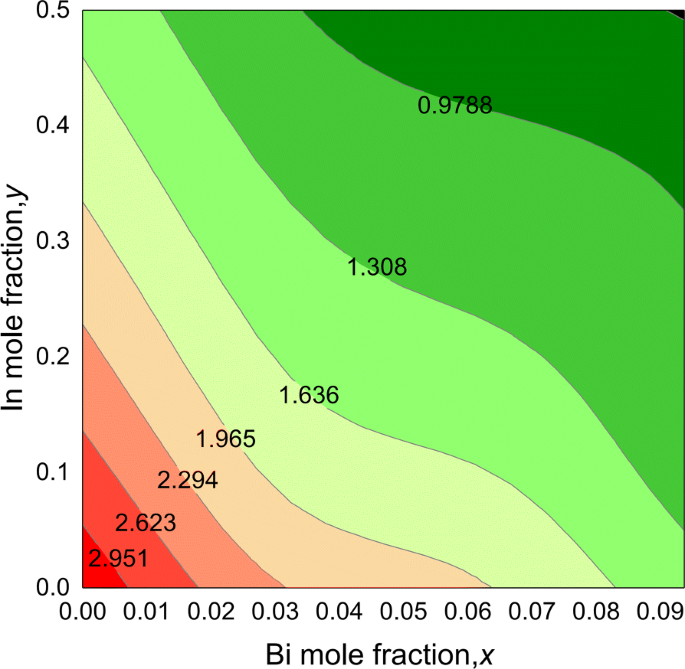
Gráfico de contorno de los valores de banda prohibida para I n y G a 1− y N 1− x B yo x aleaciones, en función de Bi ( x ) y en ( y ) composiciones
En comparación con InGaN, la incorporación de Bi induce una reducción más nítida de la banda prohibida. Pero más allá de eso, un aumento significativo en Δ SO se obtiene debido al fuerte efecto SOC del bismuto donde la interacción avanzada entre el espín del electrón y el momento angular orbital disminuye la energía de la banda SO. Además, el borde mejorado de la banda de valencia surgido del efecto anti-cruzamiento de la banda de valencia de las aleaciones de bismido también mejora en gran medida Δ SO [28]. Nuestro Δ calculado SO los valores son aproximadamente 0,220 eV para 3,125%, 0,360 eV para 6,25% y 0,600 eV para 9,375% de Bi, respectivamente, lo que tiene una variación insignificante con la fracción de indio. Investigaciones anteriores han demostrado que las diferentes disposiciones de Bi tienen una gran influencia en las estructuras de bandas de las aleaciones de bismido, incluida la energía de división de la órbita de espín [21, 52]. Los resultados actuales muestran que el I n 0.5 G a N B yo 0.09375 El valor de la banda prohibida (0,651 eV) es muy cercano al de Δ SO (0,577 eV). Dado que la muestra de InGaN presenta un gran desajuste de celosía y una calidad deficiente para un contenido de In superior al 55-60% [32], así como la baja solubilidad del bismuto en las aleaciones de bismuro diluido, establecemos los contenidos de In hasta un 50% y Bi hasta 9,375%. Creemos que un mayor contenido de indio o bismuto logrará Δ SO > E g en la muestra cuaternaria de InGaNBi para mejorar la eficiencia de los LED y LD basados en InGaNBi.
Las estructuras de bandas proyectadas y la densidad total de estados (TDOS) de GaN prístino, I n 0,25 GaN y I n 0,25 G a N B yo 0.03125 las aleaciones se presentan en la Fig. 5. Las contribuciones de In y Bi están resaltadas por color:azul (rojo) corresponde al estado que se origina en In (Bi). La sustitución de In en I n 0,25 GaN tiene una gran influencia tanto en la banda de conducción como en la banda de valencia, donde el mínimo de la banda de conducción (CBM) se empuja a energías más bajas con respecto al nivel de Fermi y refleja una brecha de energía más estrecha. A diferencia del bismuto que introduce la banda de defecto en el espacio prohibido cerca del nivel de Fermi, los átomos In muestran una hibridación con el nivel profundo del VB. Para aleación cuaternaria I n 0,25 G a N B yo 0.03125 , se puede ver claramente que la reducción de la banda prohibida resulta tanto del máximo de la banda de valencia hacia arriba (VBM) como del CBM hacia abajo, y el CBM cambia de manera más significativa en comparación con I n 0,25 GaN, que se atribuye a una mayor deformación por compresión en InGaNBi por la adición de bismuto. El nivel de defecto resaltado por el color rojo tiene una fuerte interacción con el borde VB, que se deriva de la hibridación principalmente entre los átomos de Bi y Ga cercanos. El TDOS en la Fig. 5e también refleja el nivel de defecto local en −1.0 a −0.5 eV.
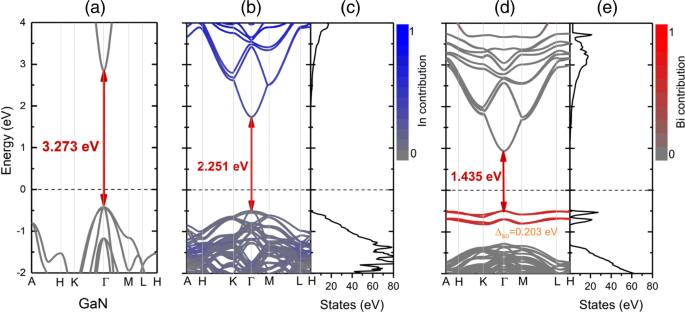
Las estructuras de bandas proyectadas y su correspondiente densidad total de estados (TDOS) de a GaN, b , c yo n 0,25 G a N y d , e yo n 0,25 G a N B yo 0.03125 . La línea punteada negra representa el nivel de Fermi, que se establece en cero. Las contribuciones relativas de In y Bi están resaltadas por color:azul (rojo) corresponde al estado que se origina en In (Bi)
Cepa de InGaNBi en GaN
I orientado a [0001] n y G a 1− y Los pozos cuánticos con tensión de N / GaN se adoptan ampliamente en los dispositivos LED y LD actuales, en los que I n y G a 1− y N capas sufren una tensión de compresión biaxial. Las fluctuaciones de composición locales y los diferentes radios covalentes de átomos de In y Ga dan lugar a las deformaciones en I n y G a 1− y N capas [53]. La Figura 6 muestra la cepa de InGaNBi sobre un sustrato de GaN. Dado que el átomo de indio es más grande que el átomo de galio, el átomo de bismuto es más grande que el átomo de nitrógeno; por tanto, la incorporación de átomos de In y Bi en InGaNBi induce una deformación compresiva de InGaNBi en GaN. Se muestra que en el contenido de In del 50% y el contenido de Bi del 9,375%, InGaNBi se encuentra bajo una alta tensión de compresión del 8,5%. Para la fracción In dentro del 6.25% y la fracción Bi dentro del 2.8%, la deformación de InGaNBi en GaN está dentro del 1%. Es decir, mediante el ajuste de las composiciones de In y Bi, InGaNBi se puede diseñar en GaN con una cepa aceptable.
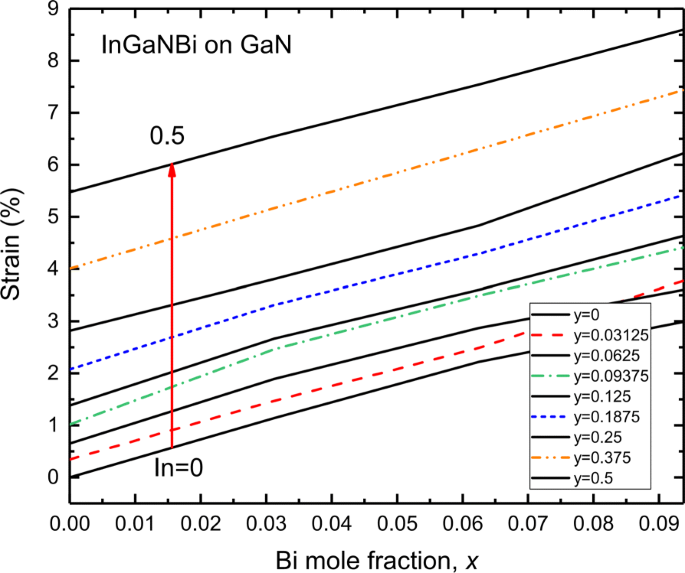
Deformación de las aleaciones de InGaNBi sobre el sustrato de GaN a varios In (0–0,5) en función de la fracción Bi. Los valores positivos de tensión indican que InGaNBi está bajo tensión de compresión
Conclusiones
Las propiedades estructurales, electrónicas y la tensión de InGaNBi en composiciones de GaN frente a In y Bi se investigan basándose en la teoría funcional de la densidad. Los parámetros de la red de InGaNBi aumentan casi linealmente con el aumento de las composiciones de In y Bi. Dado que los átomos de In y Bi tienen el radio atómico más grande que el de los átomos de Ga y N, las longitudes de los enlaces In-N y Ga-Bi son mayores que las de Ga-N. Para las propiedades electrónicas, hemos mostrado el gráfico de contorno para la banda prohibida del I cuaternario n y G a 1− y N 1− x B yo x aleaciones. La banda prohibida de las aleaciones cuaternarias podría cubrir un amplio rango de energía de 3.273 a 0.651 eV para Bi hasta 9.375% e In hasta 50%, correspondiente al rango de longitud de onda de 0.38 a 1.9 µm. El Δ calculado SO los valores son aproximadamente 0,220 eV para 3,125%, 0,360 eV para 6,25% y 0,600 eV para 9,375% de Bi, respectivamente, lo que tiene una variación insignificante con la fracción de indio. Creemos que una composición más alta de indio o bismuto logrará Δ SO > E g en la muestra cuaternaria de InGaNBi para mejorar la eficiencia de los LED y LD basados en InGaNBi. Los análisis de la estructura de la banda muestran que el indio tiene una gran influencia tanto en CB como en VB, y el bismuto tiene una fuerte interacción con el borde VB. Finalmente, investigamos la cepa de InGaNBi en GaN. Mediante el ajuste de las composiciones de In y Bi, InGaNBi se puede diseñar en GaN con una cepa aceptable.
Nanomateriales
- PPA reforzado con fibra de carbono para componentes electrónicos y automotrices estructurales
- Estructura y propiedades electrónicas de la nanoarcilla de caolinita dopada con metal de transición
- Modulación de las propiedades de anisotropía óptica y electrónica de ML-GaS por campo eléctrico vertical
- El efecto del plasma de no equilibrio por contacto sobre las propiedades estructurales y magnéticas de Mn Х Fe3 - X О4 Espinelas
- Propiedades ópticas y electrónicas de fotodiodos N + / P de silicio hiperdopado con azufre inducido por láser de femtosegundo
- Propiedades ópticas del infrarrojo cercano visible y estructural del TiO2 dopado con Cr para pigmentos fríos coloreados
- Prueba de las propiedades estructurales, electrónicas y magnéticas de Ag n V (n =1–12) Clusters
- Dependencia de la toxicidad de las nanopartículas en sus propiedades físicas y químicas
- Estructura electrónica y características I-V de las nanocintas InSe
- Propiedades de PCB automotriz y consideraciones de diseño
- Propiedades y Composición del Arrabio