


Propiedades electrónicas de la adsorción de átomos de vanadio en una superficie limpia y cubierta de grafeno de Cu (111)
Resumen
Las propiedades electrónicas de los átomos de vanadio adsorbidos en una superficie de Cu (111) limpia y cubierta de grafeno se han estudiado sistemáticamente utilizando un método teórico ab initio. En este trabajo se consideran dos coberturas (1/9 ML y 1 ML) de adsorción de vanadio. Nuestros cálculos indican que V que permanece debajo de la superficie de Cu resulta ser el sitio de adsorción más estable en las dos coberturas antes mencionadas para V / Cu (111). Sin embargo, tal adsorción puede dar lugar a propiedades no deseadas. Por lo tanto, introducimos el grafeno como una capa amortiguadora para aliviar eficazmente la interacción directa entre la superficie de V y Cu. Los cálculos muestran que las propiedades electrónicas de la capa de grafeno original se ven significativamente afectadas por las interacciones de los átomos de C con los V adatomos; el punto de Dirac del grafeno se "destruye" como consecuencia en ambas coberturas. En el sistema V / Gra / Cu (111), la interacción entre la capa de grafeno y los átomos de Cu del sustrato permanece débil como en el sistema Gra / Cu (111). Además, una cobertura relativamente baja de 1/9 ML da lugar a un sistema de espín polarizado, mientras que se observa un sistema sin espín polarizado en la cobertura de 1 ML. Este hallazgo ofrece una nueva forma para la aplicación de materiales a base de vanadio en la realidad.
Antecedentes
La catálisis heterogénea juega un papel crucial en muchas áreas de las industrias química y energética. Los estudios intensivos se han centrado en comprender, mejorar y diseñar nuevos catalizadores hasta ahora. La adsorción de átomos de metales de transición en un sustrato de metal noble puede influir en las propiedades catalíticas correspondientes, que es uno de los temas más importantes de la catálisis [1,2,3,4,5,6,7]. En particular, la adsorción de un metal monocapa sobre la superficie del metal exhibe propiedades químicas y catalíticas significativamente diferentes dentro de varios tipos de sistemas adsorbidos [5, 6, 7]. En general, las propiedades catalíticas de los materiales dependen de sus estructuras atómicas, composición y estados electrónicos que se acercan al nivel de Fermi [8,9,10,11,12]. Se espera que el sustrato influya directa y / o indirectamente en las propiedades catalíticas de los depósitos metálicos. Como todos sabemos, la superficie de Cu (111) es una de las superficies de metal monocristalino más investigadas en las últimas décadas [13,14,15,16,17,18,19,20,21,22,23,24 ]. Especialmente, en la última década, la superficie de Cu (111) ha sido considerada como el sustrato más primario para el crecimiento de grafeno de alta calidad y área grande por deposición química de vapor (CVD) [22,23,24]. Las nuevas propiedades electrónicas del grafeno se pueden conservar bien en un sustrato de este tipo. La adsorción de metales de transición 4d tardíos (como Rh [25], Pd [26,27,28,29,30], Ir [31] y Pt [29, 32, 33]) en la superficie de Cu (111) han sido ampliamente estudiados tanto experimental como teóricamente. Sin embargo, el estudio de los primeros átomos de metales de transición 3d adsorbidos en la superficie de Cu (111) es relativamente escaso [34,35,36,37]. Aquí, nos enfocamos en el elemento de metal de transición 3d temprano, vanadio, debido a su relevancia bioquímica y extensas aplicaciones en varios campos industriales, como catálisis heterogénea, redes moleculares, nanomateriales y construcción de baterías [38]. Se propone que los materiales de polianión a base de vanadio sean los candidatos para reemplazar los materiales de cátodo comerciales LiCoO 2 y LiMn 2 O 4 debido a sus estados de valencia flexibles [39]. Por tanto, estudiar las características de adsorción de los átomos de vanadio puede facilitar sus aplicaciones en la realidad. Las posibles aplicaciones de los sistemas estudiados se pueden esperar a continuación. (1) Los estados de oxidación comunes del vanadio pueden ser + 2, + 3, + 4 y + 5; por lo tanto, se puede utilizar como un catalizador potente y versátil en la industria de los nanomateriales [38]. (2) El vanadio en estado metálico se puede utilizar para catalizar la desproporción de CO a C y CO 2 [40]. (3) También es interesante analizar los átomos de TM (es decir, vanadio) adsorbidos en superficies con una concentración débil de electrones libres debido al posible aumento de la conducción eléctrica y térmica [41]. Además, existe un interés extraordinario en el orden magnético en sistemas de superficies bidimensionales que se pueden utilizar en medios de grabación, tintas magnéticas y dispositivos espintrónicos.
En este trabajo, presentamos una investigación sistemática de la adsorción de átomos de vanadio en una superficie limpia de Cu (111) y en una superficie de Cu (111) cubierta de grafeno basada en la teoría funcional de la densidad (DFT). Para los dos sistemas antes mencionados, se consideran dos coberturas contrastivas (es decir, 1/9 ML y 1 ML) de los adatomos de vanadio para evaluar el efecto de la cobertura sobre las propiedades electrónicas y magnéticas. El sitio de adsorción de energía más baja para V adsorbido en la superficie limpia de Cu (111) está debajo de la superficie en lugar de por encima de la superficie, independientemente de las coberturas de V. Para la adsorción de V en la superficie de Cu (111) cubierta de grafeno, los sitios de adsorción dependen de la cobertura, es decir, el sitio hueco con máxima coordinación se ve favorecido energéticamente para una cobertura de 1/9 ML, mientras que el sitio superior con baja coordinación se prefiere para una cobertura de 1 ML. Mientras tanto, para los sistemas V / Cu (111) y V / Gra / Cu (111), las polarizaciones de espín de los V adatomos se favorecen energéticamente para la cobertura de 1/9 ML mientras que no se encuentra magnetismo para la cobertura de 1 ML. Además, un momento magnético neto para los átomos de C de grafeno es de aproximadamente 0,16 μ B / por carbón para el sistema 1/9 ML V / Gra / Cu (111), que es diferente a los resultados en el sistema Gra / Cu (111). Para obtener una comprensión profunda de las interacciones en los sistemas V / Cu (111) y V / grafeno / Cu (111), se analizan en detalle los estados electrónicos en la superficie de Fermi. En resumen, nuestros estudios podrían ayudar a comprender las propiedades electrónicas de los sistemas V / Cu (111) y V / Gra / Cu (111).
Métodos
Nuestros cálculos se han realizado empleando el paquete de simulación ab initio de Viena (VASP) [42] que se basa en la teoría funcional de densidad de espín polarizado [43], la base de onda plana y la representación de onda aumentada del proyector (PAW) [44 ]. En los cálculos se emplea la función de energía de correlación de intercambio de Perdew-Burke-Ernzerhof (PBE) [45] dentro de la aproximación de gradiente generalizado (GGA) (algunos estudios comparativos que utilizan funciones híbridas B3LYP [46, 47] y HSE06 [48] también son presentado cuando sea necesario). Para describir con precisión las interacciones de van der Waals (vdWs) entre el grafeno y la superficie de Cu (111), se adopta el PBE funcional con la corrección de vdWs (DFT-D2) [49]. La energía cinética de onda plana de corte se establece en 500 eV. La superficie de Cu (111) se modeló utilizando un modelo de placa que comprende siete capas de Cu junto con un espacio de vacío de aproximadamente 20 Å. Se modelaron diferentes coberturas de V en superficies de Cu (111) y Gra / Cu (111) utilizando diferentes supercélulas. Para las coberturas de 1/9 ML y 1 ML de V, empleamos (3 × 3) y (1 × 1) celdas unitarias de superficie, respectivamente. El esquema Monkhorst-Pack [50] con 24 × 24 × 1 k - se utilizó una malla para muestrear la integración de la zona de Brillouin para la celda unitaria de superficie (1 × 1), mientras que una celda de unidad de superficie de 8 × 8 × 1 k - Se utilizó malla para la celda unitaria de superficie (3 × 3). Durante la optimización, las tres capas de Cu más bajas de la losa se congelaron mientras que los átomos restantes de los sistemas se relajaron por completo hasta que la fuerza en cada átomo fue inferior a 0,01 eV / Å. Los átomos de vanadio se adsorbieron en un lado de la losa. La corrección dipolo [51] no se considera en este estudio debido a una corrección de energía insignificante encontrada sobre la base de nuestros cálculos.
Resultados y discusión
Adsorción de átomos de vanadio en una superficie limpia de Cu (111)
En esta sección, presentamos los resultados de la adsorción de V directamente sobre la superficie limpia de Cu (111) en dos coberturas (es decir, 1/9 ML y 1 ML). Para encontrar un sitio de adsorción favorable del átomo de V en la superficie de Cu (111), se consideran siete posibles sitios de adsorción para cada cobertura, es decir, el sitio superior, fcc, hcp, subT, top-fcc, top-hcp y puente. , como se muestra en la Fig. 1a. En particular, cabe señalar que el subT es un sitio debajo de la superficie de Cu, donde V adatom intercambia su posición con el átomo de Cu de la capa superficial (y mueve el átomo de Cu al sitio directamente encima del átomo de vanadio); ver Fig. 2a, b. Para ambas coberturas de adsorción, las energías de adsorción para los siete sitios de adsorción se calculan para el sistema V / Cu (111). Los resultados obtenidos se dan en la Fig. 1b, c. Aquí, la energía de adsorción por átomo de vanadio (E ad ) se calcula mediante la siguiente fórmula:
$$ {\ mathrm {E}} _ {\ mathrm {ad}} =\ left [\ left ({NE} _V + {E} _ {Cu (111)} \ right) - {E} _ {V / Cu (111)} \ derecha] / N $$donde E V es la energía de un átomo de vanadio aislado, E Cu (111) es la energía total de una superficie limpia de Cu (111) involucrada, E V / Cu (111) es la energía total de un sistema V / Cu (111), y N es el número de átomos de V involucrados. De la Fig. 1b, c, se puede ver que los sitios subT se favorecen energéticamente para las adsorciones de V en la superficie de Cu (111) para las coberturas antes mencionadas. En base a esto, consideraremos solo los sitios subT en las siguientes discusiones. Las energías de adsorción calculadas, las longitudes de enlace entre el átomo de V y sus átomos de Cu adyacentes y los momentos magnéticos atómicos de los V adatomos para el V / Cu (111) se enumeran en la Tabla 1. Como podemos ver en la Tabla 1, las energías de adsorción E anuncio son 2,17 y 1,61 eV por átomo de V para coberturas de 1/9 ML y 1 ML, respectivamente, lo que indica que la interacción de los átomos V con la superficie de Cu (111) es bastante fuerte. Además, la energía de adsorción se reduce con el aumento de la cobertura V, lo que significa que las interacciones V-V se vuelven más fuertes mientras que las interacciones entre la capa V y la superficie de Cu se debilitan. Las longitudes de enlace más cortas entre el átomo de V y sus átomos de Cu adyacentes son 2,27 y 2,37 Å para coberturas de 1/9 ML y 1 ML, respectivamente. Esto implica que la interacción entre el V adatom y el sustrato de Cu es relativamente más fuerte para 1/9 ML, lo que concuerda con los resultados calculados de la energía de adsorción. El orden ferromagnético (FM) de los V adatomos también se considera en los cálculos, y la energía de polarización de espín del orden FM se calcula mediante Δ E =( E no _ mag - E FM ) / N (con E no _ mag es la energía del estado no magnético). La energía de polarización de espín de un átomo de vanadio es 110 meV para la cobertura de 1/9 ML (ver Tabla 1), mientras que no hay magnetismo para la cobertura de 1 ML. El momento magnético atómico de V es 1,34 μ B para la cobertura de 1/9 ML de vanadio, que es muy diferente del valor (3 μ B ) de un átomo de V en fase gaseosa. Discutiremos sobre este punto más adelante.
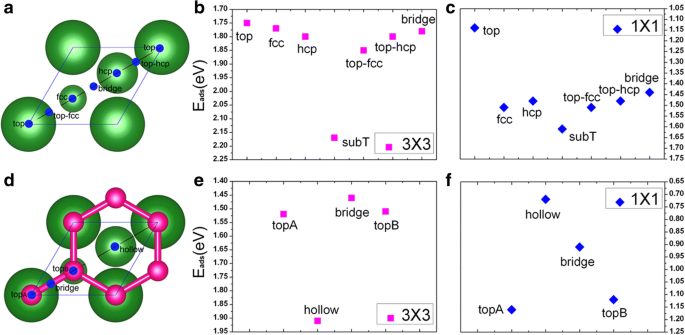
un Sitios de adsorción en una superficie limpia de Cu (111) (1 × 1):las bolas más grandes indican los átomos de Cu en la superficie y las bolas más pequeñas indican los átomos de Cu de la subcapa; b , c energías de adsorción de diferentes sitios de adsorción de átomos de V en Cu (111) en coberturas de 1/9 ML y 1 ML, respectivamente; d una superficie de Cu (111) (1 × 1) cubierta de grafeno, las bolas rojas indican átomos de C de grafeno; e, f energías de adsorción de átomos de V en Cu (111) cubierto de grafeno en coberturas de 1/9 ML y 1 ML, respectivamente
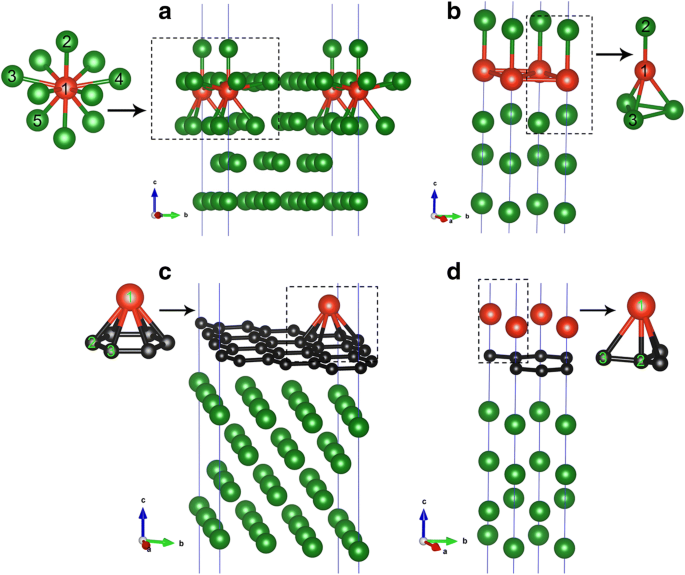
Geometrías de sistemas V / Cu (111) para a 1/9 ML y b Coberturas de 1 ML. Geometrías de sistemas V / Gra / Cu (111) para c 1/9 ML y d Cobertura de 1 ML de átomos de V. Las bolas rojas, negras y verdes representan átomos de V, C y Cu, respectivamente
A continuación, discutimos las estructuras electrónicas de los sistemas V / Cu (111). Las estructuras de bandas de las adsorciones de V en las superficies de Cu (111) (3 × 3) y Cu (111) (1 × 1) (es decir, 1/9 y 1 ML) se presentan en la Fig.3, y las estructuras de las bandas del Las superficies limpias correspondientes de Cu (111) (3 × 3) y Cu (111) (1 × 1) también se representan para comparación. Tanto la Fig. 3a, como la d se pueden utilizar para discutir las estructuras electrónicas de un Cu limpio (111); elegimos aquí la Fig. 3d. Descubrimos que ambos s electrones y d los electrones de Cu contribuyen a la conductancia del sistema para la superficie limpia de Cu (111). Más detalladamente, etiquetamos los puntos representativos (A, B, C, D, E) en la superficie de Fermi en la Fig. 3d. Los puntos A y E provienen principalmente del d yz electrones de los átomos de Cu de la superficie. Los puntos B y C muestran las contribuciones del d xy y d x 2 - y 2 electrones de átomos de Cu, respectivamente. El punto D describe la mezcla de s electrones con d z 2 y d x 2 - y 2 electrones entre los átomos de Cu vecinos. Cuando V se adsorbe en el Cu (111), las estructuras de bandas obtenidas cambian de forma diferente a medida que varía la cobertura de V. Para una cobertura de 1/9 ML (mostrada en la Fig. 3b, c), las estructuras de banda en los canales de spin-up y spin-down son diferentes, lo que indica una característica de spin-polarized. En la Fig. 3, los puntos rojos representan las contribuciones de los V adatomos, mientras que los puntos grises plateados muestran las contribuciones del Cu de fondo. Desde el canal de spin-up (es decir, spin-up mayoritario), ambos d Los electrones de los V adatomos y los átomos de Cu del sustrato contribuyen significativamente a los estados electrónicos en la superficie de Fermi. Las hibridaciones del d los electrones de los átomos de Cu de superficie y los V adatomos son claramente visibles. Para describirlo claramente, también etiquetamos algunos puntos representativos (A, B, C, D) en la superficie de Fermi en la Fig. 3b. Allí, el punto A muestra la mezcla de d z 2 electrones de V adatomos con d yz , d z 2 electrones de los átomos de Cu de la superficie. Todos los puntos B, C y D indican las contribuciones del d electrones de V adatomos. Como ejemplo, el punto B demuestra la contribución de solo el d x 2 -y 2 electrones de V adatomos. Para el canal de giro hacia abajo (es decir, giro minoritario), las estructuras de banda muestran que los estados electrónicos que contribuyeron desde V adatoms están todos muy por encima del nivel de Fermi (desocupado). La contribución a la superficie de Fermi proviene principalmente de s , d electrones de átomos de Cu, con contribuciones bastante menores de los electrones de V adatomos. La diferencia existente entre estos dos canales de espín indica un momento magnético en V adatoms (1,34 μ B ). Para una cobertura de 1 ML (mostrada en la Fig. 3e, f), a diferencia de la situación en 1/9 ML, el sistema adsorbido no tiene polarización de espín. Por conveniencia, también etiquetamos los puntos representativos (A, B, C, D, E, F, G, H) en la superficie de Fermi en la Fig. 3e. Tanto los puntos A como H corresponden a las contribuciones de d yz electrones de las capas inferiores de los átomos de Cu. Los estados electrónicos en los puntos B, D, E, F y G son aportados por d electrones de V adatom. Por ejemplo, solo d x 2 - y 2 y d xz de V adatoms contribuyen a los estados electrónicos en B y D, respectivamente. Para esos puntos entre B y D en la superficie de Fermi, encontramos una mezcla compleja de d electrones de V adatomos con s, d electrones de los átomos de Cu alrededor de los átomos de V. Por ejemplo, para el punto C, las estructuras electrónicas se caracterizan por la mezcla de s, d yz , y d z 2 electrones de la capa superficial Cu y los átomos de la subcapa superior Cu con el d z 2 electrones de V adatomos.
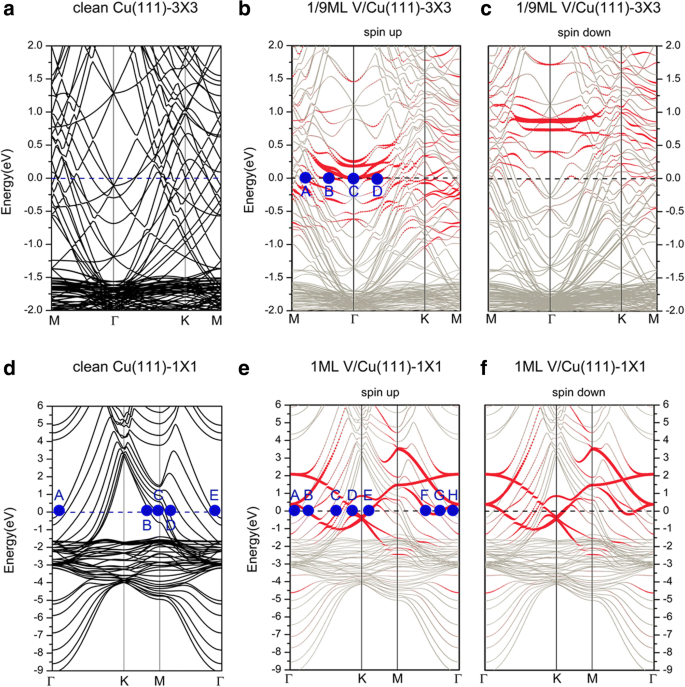
Estructuras de bandas de una superficie limpia de Cu (111), trazadas en las zonas de Brillouin (BZ) de a Celda unitaria de 3 × 3 y d 1 × 1 celda unitaria. Estructuras de bandas de adsorción de V en una superficie de Cu (111) con una cobertura de 1/9 ML para b girar y c centrifugar. Estructuras de bandas de adsorción de V en superficie de Cu (111) con cobertura de 1 ML para e girar y f girar hacia abajo
La densidad total de estados (TDOS) de adsorción de V en la superficie limpia de Cu (111), junto con la densidad proyectada de estados (PDOS), se muestran en la Figura 4 para las coberturas de 1/9 ML y 1 ML. Claramente, se encuentra una polarización de espín obvia de V adatoms a 1/9 ML (ver Fig. 4c) mientras que no se encuentra polarización de espín de V adatoms a 1 ML (ver Fig. 4f). Además, no se observa polarización de espín para los átomos de Cu en las coberturas de 1/9 ML y 1 ML (ver Fig. 4b, e). Al integrar el PDOS de Cu antes y después de la adsorción de V (es decir, lo que lleva al número de electrones en el Cu), encontramos que la carga de los átomos de Cu aumenta ligeramente, lo que indica una transferencia de carga de V adatom al sustrato de Cu en V / Cu (111). En otras palabras, la adsorción de V conduce a un dopaje de tipo n en Cu. Para comprender mejor la adsorción de V en la superficie de Cu (111), graficamos las figuras de contorno de las densidades de carga de deformación en coberturas de 1/9 ML y 1 ML en la Fig. 5a, b, respectivamente. Las densidades de carga de deformación se definen por \ (\ Delta \ rho \ left (\ overrightarrow {r} \ right) ={\ rho} _ {\ left [V / Cu (111) \ right]} \ left (\ overrightarrow { r} \ right) - \ sum \ limits _ {\ mu =1} ^ N {\ rho} ^ {atom} \ left (\ overrightarrow {r} - \ overrightarrow {R _ {\ mu}} \ right) \). Como se muestra en la Fig. 5, los enlaces covalentes e iónicos entre V adatom y sus átomos de Cu adyacentes son claramente visibles, tanto para las coberturas de 1/9 ML como de 1 ML. Específicamente, la unión covalente es relativamente más fuerte para una cobertura de 1/9 ML (en comparación con 1 ML), mientras que la unión iónica es relativamente más fuerte para una cobertura de 1 ML.
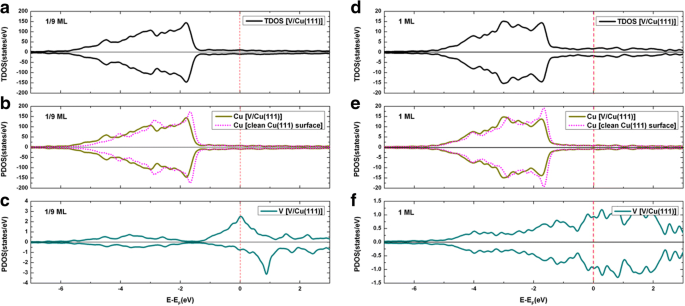
un TDOS del sistema V / Cu (111) con cobertura de 1/9 ML; b PDOS de los átomos de Cu totales del sustrato a 1/9 ML; c PDOS para V adatom a 1/9 ML; d TDOS del sistema V / Cu (111) a 1 ML; e PDOS para todos los átomos de Cu del sustrato a 1 ML; y f PDOS para V adatoms a 1 ML. Cabe destacar que el DOS de superficie limpia Cu (111) también se implanta en las gráficas b y e para comparar
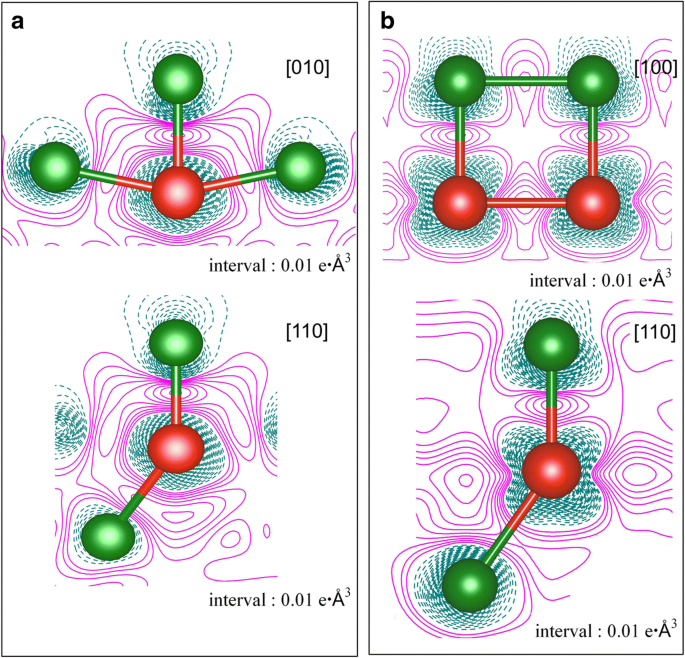
Densidades de carga de deformación para la adsorción de átomos de V en la superficie de Cu (111) en dos coberturas, es decir, a 1/9 ML y b 1 ml. La acumulación y el agotamiento de electrones están representados por líneas continuas magenta y líneas discontinuas de color cian oscuro, respectivamente. Las bolas verdes y rojas representan átomos de Cu y V, respectivamente
Capa de grafeno en la superficie de Cu (111)
Se encuentra que el sitio subT es el sitio de adsorción más estable para los átomos de V en el Cu (111) limpio de la energía de adsorción en las discusiones anteriores. Aunque tal sitio de adsorción tiene algunos intereses; sin embargo, los átomos de V que permanecen debajo de la capa superficial de Cu pueden dar lugar a propiedades no deseadas que limitan su aplicación como catalizador en la superficie. Por lo tanto, para aliviar la interacción directa entre los V adatomos y la superficie de Cu (111), intentamos introducir una capa amortiguadora. El grafeno es una elección perfecta debido a la combinación de celosía más proximal con el sistema V / Cu (111), que se ha considerado en primer lugar en este trabajo.
Las adsorciones de grafeno en superficies metálicas se han estudiado intensamente en varias publicaciones anteriores [52, 53, 54]. Cuando el grafeno se adsorbe en Cu (111), se consideraron tres posibles geometrías de los sistemas grafeno / Cu (111) (denominados como Gra / Cu (111) en adelante), es decir, el grafeno estaba en el top-fcc, top-hcp y sitios de adsorción de fcc-hcp; vea la Fig. 6a – c. Según nuestros resultados, se muestra que la geometría top-fcc (Fig.6a) es la estructura energéticamente más estable, con una energía de adsorción de 47 meV por átomo de carbono, y la distancia de equilibrio entre el grafeno y la superficie de Cu (111) es 3,14. Å, que concuerda bastante bien con los estudios anteriores [52, 53, 54]. Una energía de adsorción tan baja (47 meV / C) y una gran distancia entre capas implican que la unión entre el grafeno y el Cu (111) es relativamente débil. La Figura 6d muestra las estructuras de bandas del sistema grafeno / Cu (111) con la configuración superior-fcc (Figura 6a), especialmente, los estados electrónicos aportados por el grafeno están dibujados por círculos magenta en la figura. Las estructuras de bandas de una hoja de grafeno independiente también se muestran en la Fig. 6e para comparar. Como puede verse en estas figuras, las estructuras de bandas de grafeno son muy similares entre la hoja independiente y la de la superficie de Cu (111). El punto de Dirac en K (con cruce de banda lineal) se conserva en la Fig. 6d pero con un pequeño cambio descendente cuando el grafeno se adsorbe en la superficie de Cu (111). El cambio descendente del punto de cruce indica una transferencia de carga desde el sustrato de Cu (111) a la capa de grafeno, que está de acuerdo con los resultados anteriores de que Al, Ag y Cu son dopados de tipo n por grafeno [52,53,54 ]. Las densidades de carga de deformación, es decir, las diferencias de carga entre la densidad de carga total de Gra / Cu (111) y la suma de las densidades de carga del grafeno independiente y la superficie limpia de Cu (111), es decir, \ (\ Delta \ rho \ izquierda (\ overrightarrow {r} \ right) ={\ rho} _ {Gra / Cu (111)} \ left (\ overrightarrow {r} \ right) - {\ rho} _ {Gra} \ left (\ overrightarrow { r} \ right) - {\ rho} _ {Cu (111)} \ left (\ overrightarrow {r} \ right) \), se grafican en la Fig. 6f. Como se muestra en la Fig. 6f, también podemos observar la transferencia de carga desde el sustrato de Cu (111) a la capa de grafeno de acuerdo con las líneas de contorno sólidas alrededor de los átomos de C. Mientras tanto, trazamos la densidad proyectada de estados (PDOS) para átomos de C y Cu en el sistema Gra / Cu (111), junto con el cambio de función de trabajo de la superficie de Cu (111) (con y sin adsorción de grafeno) en la Fig. 7. A partir de la integral de la PDOS, encontramos que los electrones en los átomos de C aumentan ligeramente, mientras que los electrones disminuyen ligeramente para los átomos de Cu, lo que también verifica el fenómeno de transferencia de carga. Además, encontramos que la función de trabajo calculada de Cu (111) cambia de 4,78 a 4,68 eV después de la adsorción de la capa de grafeno. Todos estos confirman que la transferencia de carga es del sustrato de Cu a la capa de grafeno.
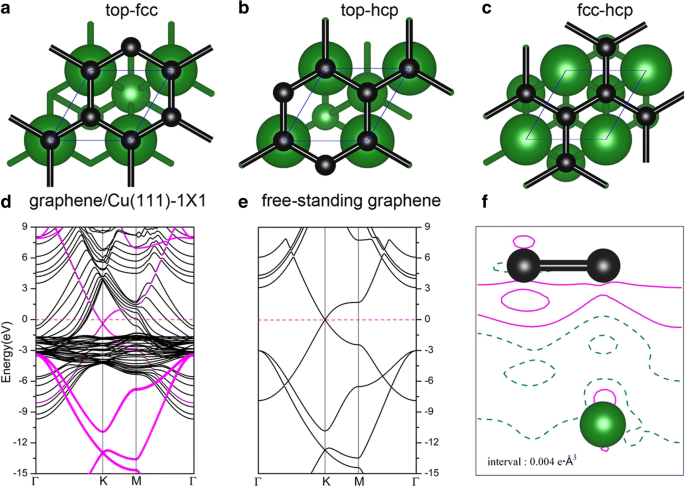
Geometría de la hoja de grafeno en la superficie de Cu (111): a top-fcc, b top-hcp y c sitios fcc-hcp. d Estructuras de bandas del sistema de grafeno / Cu (111) en la geometría superior fcc, en comparación con e los del grafeno independiente. Los puntos magenta indican los estados electrónicos aportados por el grafeno. f Las diferencias de carga entre la densidad de carga total de grafeno / Cu (111) y la suma de las densidades de carga del grafeno independiente y la superficie limpia de Cu (111) para la geometría fcc superior. El intervalo de la línea de contorno es 0.004e Å −3
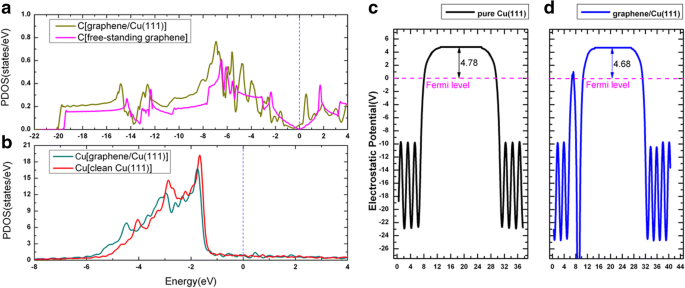
Densidad proyectada de estados de grafeno / Cu (111) para a Átomos de C y b Átomos de Cu. c, d Función de trabajo de una superficie limpia de Cu (111) y una interfaz grafeno / Cu (111), respectivamente
Adsorción de vanadio en una superficie de Cu (111) cubierta de grafeno
En esta sección, intentamos evaluar las propiedades atómicas, electrónicas y magnéticas de la adsorción de vanadio en la superficie de Cu (111) cubierta de grafeno. Se consideran cuatro posibles sitios de adsorción, etiquetados como topA, puente, topB y sitios huecos, como se muestra en la Fig. 1d. Las energías de adsorción de los átomos de V en el Gra / Cu (111) a 1/9 ML y 1 ML se dan en la Fig. 1e, f, respectivamente. El sitio energéticamente favorecido para la adsorción de V en la superficie de Cu (111) cubierta de grafeno depende de la cobertura. Para ser más específicos, los V adatoms prefieren los sitios huecos coordinados al máximo (ver Fig.1d) para 1/9 ML, mientras que los sitios superiores con coordinación baja (es decir, el sitio topA, ver Fig.1d) se prefieren para una cobertura alta de 1 ML . Las energías de adsorción, las longitudes de enlace entre el átomo de V y sus átomos de C adyacentes, y los momentos magnéticos atómicos del vanadio y el carbono para los sistemas V / Gra / Cu (111) se enumeran en la Tabla 1. Las energías de adsorción de los átomos de V en el Superficie de Gra / Cu (111), E ad , son 1,91 y 1,16 eV por átomo de V para 1/9 ML y 1 ML, respectivamente, que se reducen en cierta medida en comparación con los de la superficie de Cu (111). Obviamente, la introducción de la capa amortiguadora de grafeno puede debilitar la interacción entre los V adatomos y la superficie de Cu (111) como esperábamos. Investigamos más a fondo la energía de polarización de espín del orden ferromagnético de V en V / Gra / Cu (111) en diferentes coberturas. La energía de polarización de espín es de 390 meV para 1/9 ML, mientras que no hay polarización de espín para 1 ML. La energía de polarización de espín es considerablemente más alta en el sistema V / Gra / Cu (111) en comparación con la del sistema V / Cu (111) (390 meV en comparación con 110 meV, ver Tabla 1). El momento magnético de V adatom en el sistema V / Gra / Cu (111) a 1/9 ML es 2,93 μ B que está cerca de 3 μ B / átomo (el valor de un átomo de V en fase gaseosa), lo que implica que los átomos de V están bien aislados y hay poca carga transferida entre el átomo de V y la capa de grafeno. Un pequeño momento magnético para el átomo de C (0,16 μ B / atom) también se encuentra.
A continuación, discutimos sobre las estructuras de bandas de adsorción de V en la superficie de Cu (111) cubierta de grafeno. La Figura 8 muestra las estructuras de bandas de la adsorción de V en la superficie de Gra / Cu (111), junto con las estructuras de bandas de un grafeno independiente representado en dos celdas unitarias diferentes. Allí, los círculos azul y rojo representan las contribuciones del grafeno y V adatom, respectivamente. En primer lugar, cabe señalar que hay muchas bandas de Cu que cruzan el nivel de Fermi, lo que indica que los átomos de Cu en el sistema contribuyen significativamente a la conductancia del sistema. Para una cobertura de 1/9 ML, las estructuras de banda (ver Fig. 8b, c) también muestran que las estructuras electrónicas del sistema están polarizadas por espín. Como se mencionó en la sección anterior, la interacción entre el grafeno y la superficie de Cu (111) es muy débil para Gra / Cu (111); en consecuencia, las bandas aportadas por el grafeno se reconocen fácilmente en las estructuras de bandas generales. Sin embargo, después de la adsorción de V en la superficie de Cu (111) cubierta de grafeno (es decir, sistema V / Gra / Cu (111)), podemos ver que el punto de Dirac del grafeno está completamente "destruido" (ver Fig. 8b) en las estructuras de banda del canal de rotación, mientras que el "punto de cruce lineal" del grafeno todavía es distinguible en el canal de rotación. Sin embargo, el cambio muy hacia abajo del "punto de cruce lineal" en el componente de giro hacia abajo indica un número relativamente grande de transferencia de carga a la capa de grafeno. Cabe señalar que las cargas transferidas a la capa de grafeno provienen de la capa de átomos de V (ver Fig. 10a), ya que las interacciones entre las capas de C y Cu son débiles. Para el canal de spin-up a 1/9 ML, encontramos que, a excepción de una gran cantidad de d Los electrones de los átomos de Cu del sustrato contribuyen a la superficie de Fermi, también hay muchos d electrones de V adatom contribución a la conductancia del sistema. Mientras tanto, la hibridación de p electrones de átomos de C con ambos d los electrones de V adatom y los átomos de Cu de la superficie son visibles (pero no notables). Asimismo, etiquetamos dos puntos representativos (A, B) en la superficie de Fermi en la Fig. 8b. El punto A representa la contribución del d xy , d x 2 - y 2 electrones de V adatom, mientras que el punto B muestra la contribución de la hibridación de p z electrones de átomos de C con el d z 2 electrones de V adatom y capa superior de átomos de Cu. Claramente, la capa de superficie V es una capa conductora importante. Por el contrario, en el canal de spin-down a 1/9 ML, los estados electrónicos en el nivel de Fermi se originan principalmente en d electrones de átomos de Cu y p z electrones de átomos de C; la contribución de d electrones de V adatom es insignificante. Para una cobertura de 1 ML, las estructuras de bandas, que se muestran en la Fig. 8e, f, implican que el sistema no está polarizado por espín. Dado que el punto de Dirac de la capa de grafeno también está "destruido", la interacción entre los V adatomos y la capa amortiguadora de grafeno debería ser fuerte. A partir de los estados electrónicos calculados del sistema, vemos que los electrones contribuidos al nivel de Fermi son principalmente de s , d electrones de Cu y d electrones de átomos de V, así como el p -electrones de átomos de C. En más detalles, los puntos k que se encuentran en la superficie de Fermi, A, B, C, D y E, están etiquetados en la Fig. 8e. El punto A indica la hibridación de d yz y d z 2 de los átomos de Cu de la primera capa (superior). Los puntos B, C y D son los estados electrónicos del d -electrones de V adatom. Más específicamente, el punto B describe los estados de los electrones de d xy , d x 2 - y 2 electrones de V adatom. Además, el punto E muestra la fuerte hibridación de s , d z 2 electrones de V adatom con p z electrones de átomos de C, junto con una mezcla relativamente más débil de p z electrones de átomos de C con d z 2 electrones de la capa superior de átomos de Cu.
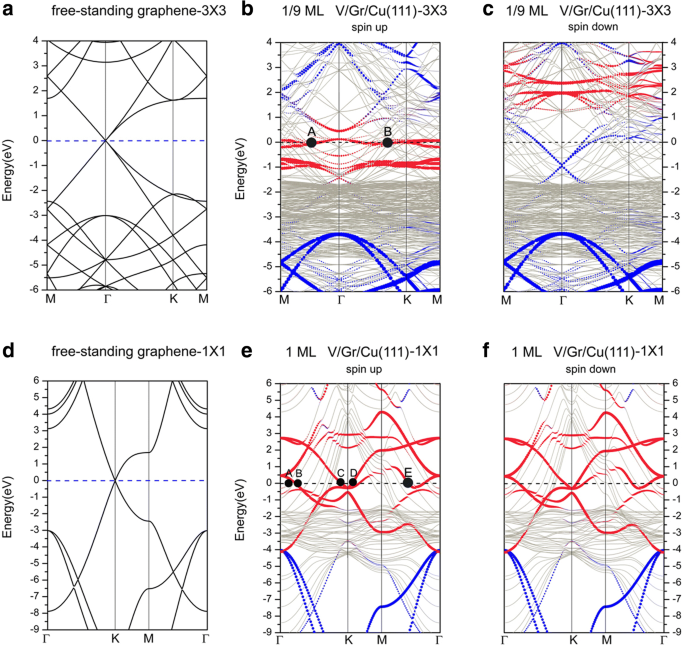
Estructuras de bandas de un grafeno independiente, trazadas en las zonas de Brillouin de a una celda unitaria (3 × 3) y d una celda unitaria (1 × 1). Estructuras de bandas de adsorciones de V en las superficies de Cu (111) cubiertas de grafeno para b centrifugado de 1/9 ML, c centrifugado de 1/9 ML, e giro de 1 ML y f spin down of 1 ML
We now present the density of states for the V/Gra/Cu(111) system. The total density of states of V adsorption on the graphene-covered Cu(111) surface, together with the projected densities of states, are demonstrated in Fig. 9 for both the 1/9 ML and 1 ML coverages. At 1/9 ML, the spin polarizations of V adatoms and C atoms in the graphene layer are clearly seen (see Fig. 9c, d), while no spin polarization is found for Cu atom (see Fig. 9b). At 1 ML coverage, no spin polarization has been found for all atoms (see Fig. 9f-h). At 1/9 ML coverage, the DOS of the spin-up channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and V atoms (totally 5.8 states/eV∙u.c.), with only minor contributions from the graphene layer (totally 0.4 states/eV∙u.c.). Meanwhile, the DOS of spin-down channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and graphene layer (totally 1.1 states/eV∙u.c.), with only minor contributions from the V atoms (totally 0.1 states/eV∙u.c.). For the 1 ML coverage, both the DOS of spin-up and spin-down channels at the Fermi level are mainly contributed from the Cu atoms and V atoms (i.e., 1.1 and 0.7 states/eV∙u.c. for each spin component, respectively), with negligible contribution from the graphene layer (0.04 states/eV∙u.c). By integrating the PDOSs for each atom before and after the V adsorptions (leading to number of electrons), the charge transfer can be determined for different atoms. To be specific, the total valence electrons of the Cu atoms are reduced slightly for both the 1/9 ML and 1 ML coverages when compared with those of a clean Cu(111) surface, while the total valence electrons of C atoms are slightly increased when compared with that of a free-standing graphene. This implies that small amount of charges are transferred from Cu substrate to graphene layer for V/graphene/Cu(111) systems regardless of the V coverages. The total valence electrons of Cu atoms in V/Gra/Cu(111) systems are almost equal to those in the graphene/Cu(111) systems, which indicates that the Cu substrate has not been affected by V adsorption. The physical pictures given by the analysis of DOSs here are all in consistent with the analysis of the band structures. Finally, we show in Fig. 10 the contour plots of the deformation charge densities for the 1/9 ML and 1 ML coverages, respectively. The deformation charge density is defined as \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Gra/ Cu(111)\right]}\left(\overrightarrow{r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \). As shown in Fig. 10, the interactions between the graphene layers and the substrate Cu atoms are both relatively weak for 1/9 ML and 1 ML coverages, which are in consistent with the above discussions. From Fig. 10a, for the 1/9 ML, the bonding between V adatoms and its adjacent C atoms is mainly ionic, and the covalent bonding is not obvious. In contrast, for the 1 ML coverage, both ionic and covalent bonding between V adatom and its adjacent C atoms are clearly visible (see Fig. 10b). Besides, the covalent bonding between neighboring V adatoms is also very significant at 1 ML coverage. Due to the existence of graphene buffer layer, V adatoms cannot interact directly with the Cu atoms.

un TDOS of V adsorption on graphene-covered Cu(111) surface at 1/9 ML coverage; PDOS for b Cu atoms, c C atoms, and d V adatoms at 1/9 ML. Likewise, e TDOS of V adsorption on Gra/Cu(111) surface at 1 ML coverage; PDOS for f Cu atoms, g C atoms, and h V adatoms at 1 ML. For comparison, PDOS of Cu atoms of a clean Cu(111) and graphene/Cu(111) are implanted in b y f , while PDOS of C atoms of a free-standing graphene are also implanted in c and g

Deformation charge densities for the adsorption of V atoms on the graphene-covered Cu(111) surface at two coverages, i.e., a 1/9 ML and b 1 ML. Electron accumulation and depletion are represented by magenta solid lines and green dashed lines, respectively. The green, black, and red balls represent Cu, C, and V atoms, respectively
We have also calculated the phonon spectra for both the V/Cu(111) and V/Gra/Cu(111) systems. From the calculated phonon spectra, we find that there is no “imaginary frequency” for both the two types of systems, indicating that the systems studied are dynamically stable and would be seen in the laboratory. Since the main purpose of our work is not the thermodynamic stability, therefore the figures of the phonon dispersions are not shown in this text. Second, we have noticed that the different DFT functionals we adopted may lead to the different results. Hence, we have calculated the 1 ML V/Gra/Cu(111) system (as a representative) within the DFT framework under the B3LYP, HSE06 hybrid functionals, as well as the PBE functional. The results suggest that the adsorption site with largest adsorption energy is the topA site, calculated from all the PBE, HSE06, and B3LYP methods. However, relative adsorption energies at different adsorption sites from the B3LYP and PBE and HSE06 methods differ significantly (results from PBE and HSE06 methods are almost the same, since this is a metallic system). On the other hand, the geometrical parameters obtained from the three functionals show good consistency. Although the detailed charge density contours are somewhat different between PBE and B3LYP method, the main bonding characteristics are the same from both the two methods. In summary, the main point is that the adsorption energies obtained from B3LYP functional are significantly larger than those from the PBE and HSE06 functionals. To explain this point, Paier et al. argued that B3LYP functional lacked of a proper description of the “free-electron-like” systems with a significant itinerant character (e.g., metals and small gap semiconductors). They have concluded that the overestimation of the total energy of the atoms can be induced by the significantly overestimation on the exchange and correlation energies of B3LYP functional. In this respect, PBE functional often shows much more reliable results [55].
Conclusiones
To summarize, using first-principles calculations, we have systematically investigated the electronic and geometric properties of the adsorption of V atoms on both the clean Cu(111) surface and the graphene-covered Cu(111) surface. Firstly, for the V/Cu(111) system, an adsorption site underneath the Cu surface layer is found as the preferable adsorption site for V atom regardless of the coverages. The hybridization of V’s d states with Cu’s d states rules the electronic properties of V/Cu(111) systems. Ferromagnetic order of V adatoms is energetically favored for 1/9 ML coverage (1.34 μB /atom), while no magnetism of V adatoms is observed for 1 ML coverage. Due to the strong interaction between V adatom and its adjacent substrate’s Cu atoms, the magnetic moment of V is significantly reduced. Secondly, the graphene/Cu(111) systems are investigated and the results agree well with the previous literatures. Thirdly, adsorptions of V on the graphene-covered Cu(111) at two coverages (i.e., 1/9 ML and 1 ML) show different preference of adsorption sites. The hollow site with maximum coordination is energetically favored for the adsorption of 1/9 ML, while the top site with low coordination is preferred for 1 ML adsorption. In V/Gra/Cu(111) systems, the interactions of C atoms with the V adatoms destroy the electronic properties of both the original graphene layer and the adsorbed atoms, represented by the strong hybridization of C’s p z -states with V adatoms’ d z 2 -states. A net magnetic moment for C atoms of graphene also appeared (0.16 μB/per carbon). In short, our study paves the way to a deep understanding of the adsorption properties of vanadium atoms on the clean Cu(111) and graphene-covered Cu(111) substrates. Simultaneously, this study also provides a reference for the possible applications of the V/Cu(111) and V/Gra/Cu(111) systems in the catalyst in nanomaterials industry, spintronic devices, and others.
Abreviaturas
- Cu(111):
-
(111) surface of copper
- FM:
-
Ferromagnetic
- GGA:
-
Aproximación de gradiente generalizada
- ML:
-
Monocapa
- PAW:
-
Projector augmented wave
- PBE:
-
Perdew-Burke-Ernzerhof
- V/Cu(111):
-
Vanadium atoms adsorbed on Cu(111) surface
- V/Gra/Cu(111):
-
Vanadium atoms adsorbed on graphene-covered Cu(111) surface
- VASP:
-
Vienna ab initio simulation package
- vdWs:
-
van der Waals interactions
Nanomateriales
- Preparación y propiedades magnéticas de nanopartículas de espinela de FeMn2O4 dopadas con cobalto
- Hacia los nanofluidos de TiO2:Parte 1:Preparación y propiedades
- Estructura y propiedades electrónicas de la nanoarcilla de caolinita dopada con metal de transición
- Modulación de las propiedades de anisotropía óptica y electrónica de ML-GaS por campo eléctrico vertical
- Formación y propiedades luminiscentes de Al2O3:nanocompuestos de SiOC en la base de nanopartículas de alúmina modificadas por feniltrimetoxisilano
- Ajuste de las morfologías de la superficie y las propiedades de las películas de ZnO mediante el diseño de la capa interfacial
- Propiedades ópticas y electrónicas de fotodiodos N + / P de silicio hiperdopado con azufre inducido por láser de femtosegundo
- Prueba de las propiedades estructurales, electrónicas y magnéticas de Ag n V (n =1–12) Clusters
- Estructura electrónica y características I-V de las nanocintas InSe
- Síntesis controlada y propiedades de adsorción selectiva de las nanohojas de Pr2CuO4:una discusión del mecanismo
- Propiedades de PCB automotriz y consideraciones de diseño