


Síntesis ionotérmica de silicio nanoporoso cristalino y su uso como materiales anódicos en baterías de iones de litio
Resumen
El silicio tiene un gran potencial como material de ánodo para baterías de iones de litio (LIB) de alto rendimiento. Este trabajo informa un enfoque fácil, de alto rendimiento y escalable para preparar silicio nanoporoso, en el que el siliciuro de magnesio comercial (Mg 2 Si) reaccionó con el líquido iónico ácido a 100 ° C y presión ambiental. El silicio obtenido consiste en una estructura porosa cristalina con una superficie BET de 450 m 2 / gy tamaño de poro de 1,27 nm. Cuando se recubre con la capa de carbono dopado con nitrógeno y se aplica como ánodo LIB, los compuestos nanoporosos de silicio-carbono obtenidos exhiben una alta eficiencia Coulombic inicial del 72,9% y poseen una capacidad específica de 1000 mA h g −1 a 1 A g −1 después de 100 ciclos. Este método de preparación no involucra recipientes de alta temperatura y presión y se puede aplicar fácilmente para la producción en masa de materiales de silicio nanoporosos para baterías de iones de litio o para otras aplicaciones.
Introducción
El rápido aumento del consumo y la alta dependencia de la energía fósil en la sociedad contemporánea han provocado una creciente sensación de malestar sobre el medio ambiente, el clima y el suministro de energía. Existe una demanda urgente de desarrollar dispositivos y sistemas de energía sostenibles, portátiles de alta energía y alta densidad de potencia para resolver el desajuste temporal de la fuente de energía y el medio ambiente para los estilos de vida modernos [1]. Las baterías recargables de iones de litio (LIB) son muy prometedoras para los dispositivos de almacenamiento de energía debido a su densidad de energía relativamente alta y su estabilidad de ciclo largo [2, 3]. Para satisfacer los crecientes requisitos de los LIB de alto rendimiento, se están desarrollando ampliamente varios materiales de electrodos de alta capacidad, como materiales carbonosos amorfos porosos [4, 5], compuestos a base de fósforo [6, 7], compuestos a base de silicio [8 ] y óxidos de metales de transición [9, 10]. Como componente vital, el silicio (Si) es uno de los materiales anódicos más impresionantes debido a su gran capacidad teórica (4200 mAh g −1 ), abundantes fuentes naturales y voltaje de absorción de Li relativamente seguro [11]. Sin embargo, la comercialización práctica a gran escala de material anódico de silicio está plagada de dos problemas intrincados. Por un lado, la enorme expansión y contracción volumétrica en los procesos de carga y descarga provocan la descomposición del material activo de silicio, un rápido deterioro irreversible de la capacidad de la batería [12]. Por otro lado, la baja electroconductividad intrínseca (1,6 × 10 −3 S / m) de silicio elemental también impide en gran medida la transferencia de electrones y disminuye la capacidad de velocidad del electrodo.
Recientemente, se han centrado esfuerzos considerables en eludir los problemas de estabilidad antes mencionados [13]. Se han diseñado un gran número de materiales de silicio nanoestructurados, incluidos nanotubos [14], nanocables / nanovarillas [15, 16] y nanohojas [17, 18, 19] para lograr una mayor integridad estructural y rendimiento del ciclo. Además, la preparación de compuestos porosos a base de Si también se considera un método eficaz, porque los espacios porosos apropiados en los compuestos de silicio poroso podrían actuar como amortiguadores para mitigar la expansión de volumen y, por lo tanto, mejorar el rendimiento cíclico en LIB [20, 21]. Por ejemplo, Kim et al. fabricó partículas de silicio porosas tridimensionales mediante recocido térmico y grabado en geles de Si recubiertos de butilo y SiO 2 nanopartículas a 900 ° C bajo una atmósfera de Ar, que exhibieron una capacidad estable de más de 2800 mA h g −1 después de 100 ciclos a 1 ° C [22]. An et al. informó de una vía ecológica, escalable y controlable para preparar silicio nanoporoso (NP-Si) con excelentes propiedades electroquímicas de Mg 2 comercial Aleación de Si mediante destilación al vacío a alta temperatura [23]. Aunque se han demostrado enormes avances en el rendimiento electroquímico consumado, la mayoría de los métodos de preparación para estas estructuras nanoporosas de Si son generalmente demasiado complicados para escalar.
Otra táctica eficaz para impulsar el rendimiento electroquímico del ánodo de silicio es recubrir con carbón eléctricamente conductor partículas de nanosilicio para formar nanocompuestos de silicio-carbono [19, 24], como concha de yema [25], sandía [26] y estructuras huecas [ 27]. Por ejemplo, Pan et al. diseñaron nanocompuestos de Si-C con estructura de cáscara de yema con alta capacidad específica y buena estabilidad cíclica mediante un método simple y de bajo costo basado en la tecnología de grabado con NaOH [28]. Chen y col. desarrolló un Si / B 4 estructurado con núcleo-capa C compuesto con revestimiento de grafito y demostró que dichos compuestos poseían una buena estabilidad cíclica a largo plazo [29]. Varios estudios demostraron que el carbono conductor no solo podría compensar la baja conductividad eléctrica del silicio, sino que también podría servir como un intermediario elástico para retardar el gran cambio de volumen y evitar el contacto directo entre los materiales activos de silicio y el electrolito, lo que conduce a una estabilidad cíclica mejorada. [30].
Hasta la fecha, las rutas sintéticas a las nanopartículas de silicio (NP de Si) o al silicio poroso (pSi) generalmente implican la descomposición térmica de los silanos [31], el grabado químico de las obleas de Si y la reducción magnesiotermica de SiO 2 plantillas [32, 33]. Estas preparaciones generalmente requieren varios pasos, alta temperatura, plantillas de costo relativamente alto, etc., lo que conduce a un alto costo y dificultades para escalar [34]. Recientemente, también se ha prestado mucha atención a la preparación de NP de Si en solución [35, 36]. Por ejemplo, Kauzlarich et al. informó que SiCl 4 reaccionó con NaSi o KSi en disolventes orgánicos para obtener nanopartículas de silicio [37]. Liang y col. preparó las nanoesferas de silicio en forma de nido mediante una reacción solvotermal, en la que NaSi reaccionó con NH 4 Br en el disolvente mixto de piridina y dimetoxietano en un autoclave a 80 ° C durante 24 h [38]. La síntesis de solución informada generalmente involucró agentes reductores altamente activos como metales alcalinos, LiAlH 4 , y NaSi y a menudo producían bajos rendimientos o pequeñas cantidades de NP de Si. En este sentido, para la fabricación masiva de nanosilicio, sigue siendo imperativo un enfoque simple, escalable y de bajo costo. A continuación, presentamos una preparación conveniente y de alto rendimiento de silicio poroso por oxidación de Mg 2 Si en líquido iónico ácido a 100 ° C y presión ambiental. Cuando se recubre con una capa de carbono dopado con nitrógeno y sirvió como ánodo de la batería de iones de litio, los compuestos nanoporosos de silicio-carbono obtenidos exhibieron una alta eficiencia Coulombic inicial (CE) del 72,9% y entregaron una capacidad específica de 1000 mA h g
Métodos
Materiales
El cloruro de 1-butil-3-metilimidazolio ([Bmim] Cl) fue proporcionado por Shanghai Cheng Jie Chemical Co. LTD. Cloruro de aluminio (AlCl 3 ) se adquirió de Sinopharm Chemical Reagent Co., Ltd. Siliciuro de magnesio (Mg 2 Si) y polvo de silicio comercial (1-5 μm) se compraron a Alfa Aesar. Carbonato de etileno (EC), carbonato de dietilo (DEC), carbonato de fluoroetileno (FEC) y LiPF 6 aptos para baterías fueron adquiridos de Shenzhen Kejingstar Technology Ltd., China. Todos los productos químicos y reactivos se utilizaron directamente tal como se recibieron.
Síntesis de nanopartículas de silicio poroso (pSi)
En un procedimiento típico, [Bmim] Cl (1,5 g) y AlCl 3 (4,5 g) con una relación molar de ~ 1:4 se mezclaron y se cargaron en un tubo de vidrio Schlenk. Posteriormente, 500 mg de siliciuro de magnesio (Mg 2 Si) se añadieron al tubo de vidrio y se agitaron vigorosamente a 100 ° C durante 10 h. El procedimiento anterior se llevó a cabo en una caja de guantes llena de Ar. Después de enfriar, se recogió el precipitado y se lavó con ácido clorhídrico 1 M, agua destilada y etanol. Finalmente, el producto (150 mg, rendimiento del 82%) se secó al vacío para su posterior caracterización.
Síntesis de carbono dopado con nitrógeno recubierto en nanopartículas de silicio poroso (pSi @ NC)
El procedimiento de preparación se remite a la bibliografía publicada [39, 40]. En primer lugar, se dispersaron 0,1 g de las nanopartículas de silicio poroso (pSi) obtenidas en 250 ml de agua desionizada que contenía dodecilbencenosulfonato de sodio (SDBS; 5 mg) mediante ultrasonidos durante 30 min. La mezcla se agitó vigorosamente durante 1 ha temperatura ambiente. Después de eso, 200 μL de monómero de pirrol, 0,34 g de (NH 4 ) 2 S 2 O 8 y se añadieron 1,25 ml de HCl 1 M a la solución anterior. Después de agitar la mezcla en un baño de hielo / agua durante 24 h, los polvos negros formados (indicados como pSi @ PPy) se recogieron por filtración, se lavaron con agua desionizada y se secaron al vacío. Finalmente, la muestra pSi @ PPy se calentó a una velocidad de rampa de 5 ° C min −1 en un horno de tubo a 700 ° C durante 3 h en una atmósfera fluida de Ar para obtener el compuesto pSi @ NC. El contenido de carbono se estimó mediante estudios termogravimétricos.
Mediciones electroquímicas
Las propiedades electroquímicas de las nanopartículas de silicio poroso se estudiaron utilizando una media celda de moneda CR2032, en la que láminas de metal de litio sirvieron como contraelectrodos y electrodo de referencia, el pSi @ NC preparado como electrodo de trabajo y películas macroporosas de polipropileno (Celgard 2400) como separadores. y 1,0 M LiPF 6 en una mezcla 1:1 (v / v) de carbonato de etileno (EC) / carbonato de dietilo (DEC) como electrolito. Las células CR2032 se ensamblaron en una caja de guantes con atmósfera de argón (contenido de oxígeno y agua inferior a 0,1 ppm). Los electrodos del ánodo de trabajo se prepararon mezclando el compuesto pSi @ NC obtenido, el carbono super P y el alginato de sodio en una relación en peso de 70:20:10 en agua desionizada para formar una suspensión homogénea. A continuación, la suspensión se revistió sobre una hoja de Cu y se secó al vacío a 80ºC durante 12 h. La masa de carga total de los materiales activos en el electrodo fue de aproximadamente 0,5 mg cm -2 . Los ciclos de carga-descarga de las medias celdas se realizaron en un probador de baterías Neware (Shenzhen, China) en un modo de corriente constante en el rango de 0.01–1.5 V. La voltamperometría cíclica (CV) de los ánodos preparados se midió en una estación de trabajo electroquímica CHI650d (Shanghai Chenhua Instruments Inc., China), que utiliza una celda de tres electrodos con una tasa de barrido de voltaje de 0,2 mV s −1 a temperatura ambiente. La capacidad específica se calculó en base a la masa total de los compuestos pSi @ NC.
Métodos de caracterización
Las mediciones de difracción de rayos X de potencia (PXRD) se llevaron a cabo en un difractómetro de rayos X Bruker D8 ADVANCE (radiación Cu Kα, 40 kV, 40 mA, λ =1,5418 Å). La morfología y microestructura de las muestras se obtuvieron mediante microscopía electrónica de barrido (microscopio electrónico de barrido de emisión de campo de Hitachi, S-4800), y se utilizó la espectroscopia de rayos X de dispersión de energía para analizar la distribución elemental. Se registraron imágenes de microscopía electrónica de transmisión (TEM) y TEM de alta resolución en un equipo JEM-2100. Los parámetros porosos se determinaron utilizando un analizador Micromeritics ASAP 2020 a 77 K después de desgasificar la muestra a 150 ° C durante 10 h. El área de superficie específica se calculó utilizando el método de Brunauer-Emmett-Teller (BET) de múltiples puntos, y la distribución del tamaño de los poros se analizó mediante el método de la teoría funcional de la densidad (DFT) basado en los datos de adsorción. Se utilizó espectroscopia Raman (LabRAM Aramis, Horiba, equipado con un láser de longitud de onda de 633 nm) para investigar la estructura del silicio nanoporoso, que se calibró primero con una oblea de Si (520 cm −1 ). Se utilizó el espectrómetro PHI 5000 VersaProbe para las mediciones de espectroscopía de fotoelectrones de rayos X (XPS). El análisis termogravimétrico (TGA) se realizó en un analizador térmico STA449F3 (Netzche) simultáneo en una atmósfera de aire a 10 ° C min −1 de 30 a 800 ° C en flujo de aire. Las pruebas de voltametría cíclica (CV) se realizaron en una estación electroquímica CHI650d (Shanghai Chenhua Instruments Inc., China).
Resultados y discusión
La preparación de nanopartículas de silicio poroso (pSi) a partir de Mg 2 El Si en el líquido iónico se puede expresar como Reacción 1, como se muestra en el Esquema 1. Para comprender el proceso de reacción, los productos prístinos de la Reacción 1 propuesta sin ningún tratamiento de lavado fueron recolectados y analizados directamente por PXRD (Archivo adicional 1:Figura S1) . El análisis PXRD mostró que el producto prístino estaba compuesto principalmente de Si cristalino, subproducto de sales inorgánicas MgCl 2 y reactivos Mg 2 Si y AlCl 3 . En el proceso de preparación de nanopartículas de silicio porosas, se mezclaron cloruro de 1-butil-3-metilimidazol y tricloruro de aluminio en una relación molar de 1:4 para asegurar que el sistema de reacción sea ácido. Entonces, Mg 2 El Si reaccionó con el sistema ácido para formar las nanopartículas de silicio. El rendimiento de nanopartículas de silicio poroso fue superior al 82% basado en la cantidad de átomos de Si en Mg 2 Si. La reacción se llevó a cabo en un matraz, lo que proporcionó una producción en masa de pSi que se aumentó fácilmente a escala. El uso de líquido iónico [BmimCl] -AlCl 3 era necesario para la preparación de pSi. Sin AlCl 3 , la reacción de Mg 2 No se pudo producir Si con [BmimCl]. Del mismo modo, el Mg 2 Si no pudo reaccionar con AlCl 3 solo o en otros disolventes orgánicos tales como tetrahidrofurano para producir pSi. Notamos que pSi se había preparado previamente mediante la descomposición térmica de silanos o haluros de silicio a alta temperatura, o sus reacciones con agentes reductores altamente activos como metales alcalinos, LiAlH 4 y NaSi [37, 41]. El uso de Mg 2 También se conoce el Si en la preparación de silicio nanoestructurado mediante la destilación del Mg a alta temperatura [23, 42, 43]. Sin embargo, esas reacciones produjeron a menudo bajos rendimientos o pequeñas cantidades de pSi. Por el contrario, el método informado en este trabajo es aplicable para la producción en masa de pSi.
La reacción propuesta para preparar pSi
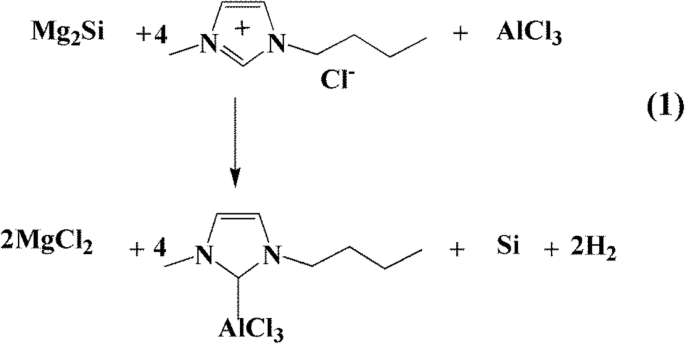
El patrón PXRD del producto se muestra en la Fig. 1a. Estos cinco picos estrechos y agudos a 2θ 28,4, 47,3, 56,1, 69,1 y 76,4 ° se asignan a los planos de celosía (111), (220), (311), (400) y (331) de la fase de silicio cúbico. (JCPDS No. 27-1402), lo que sugiere que el silicio obtenido es altamente cristalino. Los tamaños de cristalitos promedio de las partículas de silicio obtenidas fueron de aproximadamente 40 nm según la ecuación de Scherrer. La Figura 1b muestra los espectros Raman de las nanopartículas de silicio. El pico característico típico ubicado alrededor de 518 cm −1 corresponde al modo de estiramiento Si-Si del Si cristalino. La banda ancha entre 900 y 1050 cm −1 debe atribuirse al espectro de silicio de segundo orden [44]. Y el pico pequeño a ~ 303 cm −1 fue atribuido al óxido superficial. Las áreas de superficie específicas y la caracterización de la porosidad de las muestras obtenidas fueron aclaradas por N 2 isotermas de adsorción / desorción a 77 K. La muestra de pSi mostró curvas de sorción isotérmica de tipo IV (a) con un bucle de histéresis híbrido H2 (b) / H3, que es característico de un material de estructura porosa [45]. Poseía una alta superficie Brunauer-Emmett-Teller (BET) de 450 m 2 g −1 . El análisis de la distribución del tamaño de los poros basado en el método DFT mostró que el producto constaba de microporos relativamente estrechos (1,27 nm) y mesoporos (5,4 nm) con una amplia distribución del tamaño de los poros. La presencia de estos poros podría facilitar Li + difusión de iones.
un Patrones PXRD, b Espectros Raman, c Espectro XPS, d Espectro EDS, e curvas de adsorción-desorción de nitrógeno y f la curva de distribución del tamaño de los poros de pSi
La morfología de las muestras de silicio obtenidas se estudió mediante microscopía electrónica de barrido (SEM) y microscopía electrónica de transmisión (TEM). Las imágenes SEM (Fig. 2a, b, archivo adicional 1:Figura S2) y TEM (Fig. 2c, d) muestran que los tamaños generales de las partículas de las partículas de silicio nanoporoso obtenidas oscilan entre varias decenas y aproximadamente 100 nm de diámetro. La imagen TEM de la Fig. 2c muestra que la muestra está compuesta de partículas de silicio interconectadas, lo que da como resultado una estructura porosa. Postulamos que el Si 4− en el precursor de tamaño micrométrico Mg 2 Si reaccionó con líquido iónico ácido para formar Si rodeado por MgCl 2 nanopartículas. Estos últimos se lavaron con HCl diluido, dejando pSi interconectados con vacantes. El pSi obtenido mostró una gran superficie BET de 450 m 2 g −1 con una distribución uniforme del tamaño de los poros a 1,27 nm, lo que respalda la postulación anterior. La imagen HRTEM de pSi en la Fig. 2d revela que la franja de celosía transparente con un típico d -espaciamiento de 0,31 nm, atribuido a los planos cristalinos (111) del Si cúbico, concuerda bien con los resultados de PXRD. Se muestra que las nanopartículas de silicio interconectadas están cubiertas por una fina capa de óxido en la superficie exterior, debido a la oxidación. La composición de la superficie y el estado de valencia de las nanopartículas de Si se identificaron mediante análisis de dispersión de energía (EDS) y espectroscopía de fotoelectrones de rayos X (XPS). El espectro de Si 2p XPS (Fig. 1c) mostró dos picos amplios superpuestos a 98,2 eV y 103,0 eV. Los dos picos se pueden dividir en cinco componentes a 98,11, 99,11, 100,75, 102,64 y 103,64 eV, que se asignaron a Si (0), Si (I), Si (II), Si (III) y Si (IV ), respectivamente. La presencia de un pico de Si (0) fuerte implica la formación de silicio poroso. Los picos más fuertes de Si (III) y Si (IV) sugieren que la superficie del silicio poroso estaba recubierta por óxido de silicio [46]. De manera consistente, el análisis de energía dispersiva (EDS) de pSi mostró que la relación atómica de Si / O en la superficie era de aproximadamente 3:2 (Fig. 1d).

un , b Imágenes SEM y c , d Imágenes TEM de pSi (el recuadro en d muestra patrones SAED)
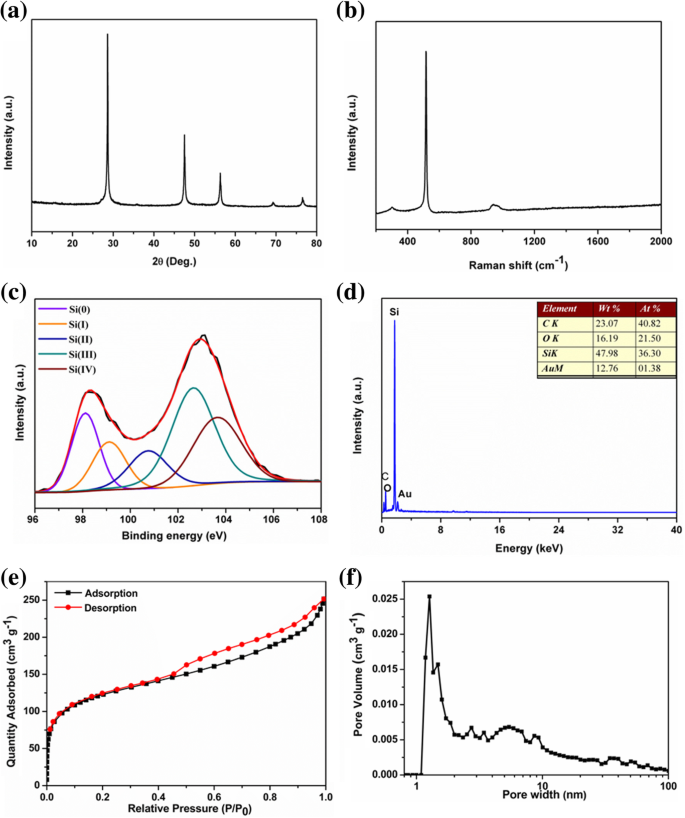
Para ser utilizados como materiales de ánodo LIB, los pSi se encapsularon con polipirrol conductor para formar compuestos pSi @ NC. El patrón PXRD del compuesto pSi @ NC mostró un pico ancho adicional alrededor de 23 ° (Fig. 3a), lo que sugiere que la capa de carbono dopado con nitrógeno es amorfa [39]. El espectro Raman del compuesto pSi @ NC (Fig. 3b) mostró dos picos anchos a 1335 y 1585 cm −1 asignado a las bandas D y G del carbono grafítico [47], respectivamente, lo que confirma el resultado de PXRD. La relación de intensidad de la banda D y la banda G (I D / I G ) del compuesto pSi @ NC es de aproximadamente 1,07, lo que implica un bajo grado de grafitización de la capa de carbono. Los espectros C 1 s XPS de pSi @ NC mostraron la existencia de enlace N-C (285,85 eV en la Fig. 3c), lo que confirma que el nitrógeno estaba dopado en la estructura de carbono [48]. El pico N 1 s XPS (Fig. 3d) se puede dividir en tres picos que se centralizan en 397,85, 398,72 y 400,57 eV, respectivamente, que pertenecen a los tipos piridínico, pirrólico y grafítico de átomos de nitrógeno dopados en la estructura de carbono [39 , 49]. El contenido de carbono en el compuesto pSi @ NC fue determinado por TGA en aproximadamente 20% en peso (Archivo adicional 1:Figura S3).
un Patrones PXRD, b Espectros Raman, c espectros XPS C 1 s de alta resolución, y d Espectros XPS de N 1 s de alta resolución de compuesto pSi @ NC
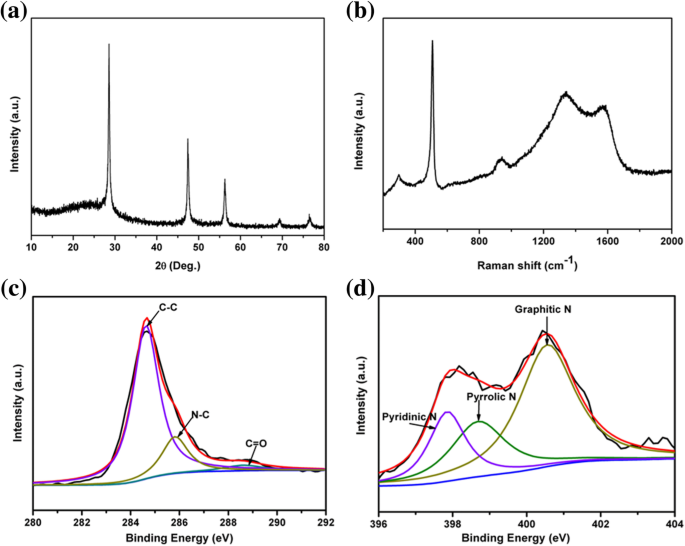
Para caracterizar el rendimiento electroquímico del compuesto pSi @ NC como el ánodo de los LIB, las mediciones de voltamperometría cíclica (CV) entre y 2,5 V a una velocidad de exploración de 0,2 mV s −1 se llevaron a cabo. Como se muestra en la Fig. 4a, el primer pico de reducción alrededor de 1,5 V en las curvas CV se atribuyó a la descomposición del aditivo electrolítico (carbonato de fluoroetileno FEC) [50]. El pico de reducción irreversible fue visible a un potencial de alrededor de 0,6 V durante la primera descarga y desapareció en los ciclos posteriores, lo que se asoció con la generación de la membrana de interfaz de electrolitos sólidos (SEI) [51]. La formación de SEI se debió a la descomposición de disolventes orgánicos de electrolitos como EC y DEC y condujo a la pérdida de capacidad inicial irreversible [50, 52]. El pico cercano a 0.1 V en las siguientes curvas CV representó la transición de silicio cristalino a Li amorfo x Si [53]. Mientras tanto, durante el proceso de carga, se observaron dos picos redox típicos de alrededor de 0,28 y 0,53 V, que estaban relacionados con el proceso de extracción de Li de Li x Si [54, 55]. En particular, las intensidades de corriente de los picos anódicos y catódicos aumentaron gradualmente después de los primeros ciclos. Este fenómeno de "activación" debe atribuirse principalmente a la degradación gradual de la estructura del silicio cristalino [54, 56].
un Curvas CV, b curvas de carga-descarga, c rendimiento de ciclismo a largo plazo a 0,1 A g −1 y 1 A g −1 durante 100 ciclos, respectivamente (densidades de corriente) y d el rendimiento de la velocidad se cicló a varias densidades de corriente del electrodo compuesto pSi @ NC. e Rendimiento cíclico del compuesto comercial Si @ NC a 0,1 A g −1 durante 100 ciclos
La Figura 4b ilustra las dos primeras curvas de descarga-carga de los ánodos compuestos pSi @ NC en ciclo a una densidad de corriente de 0.1 A g −1 . El compuesto pSi @ NC tuvo una terraza de descarga larga y plana de alrededor de 0.1 V durante la primera descarga, que está de acuerdo con la terraza característica de las inserciones de Li de Si cristalino. El silicio bien cristalizado se volvió amorfo y mostró los perfiles de carga / descarga representativos del silicio amorfo en ciclos posteriores. Las otras mesetas potenciales que aparecieron alrededor de 0,6 V durante el primer proceso de litiación fueron el resultado de la formación SEI [57]. Los resultados concordaron bien con las curvas CV. Las capacidades iniciales de carga y descarga fueron 2790 y 2036 mA h g −1 , entregando una alta eficiencia Coulombic inicial (CE) del 72,9%. La menor capacidad de carga podría deberse en parte al efecto restrictivo de la capa de óxido SiO x , que sirvieron como amortiguadores para limitar la expansión del volumen y el grado de litiación [58, 59]. Es importante destacar que no se observó una disminución obvia de la capacidad en los ciclos posteriores, y la eficiencia de Coulombic se mantuvo casi constante en alrededor del 100%.
La Figura 4c muestra el rendimiento cíclico de los ánodos compuestos pSi @ NC, que se realizaron a una densidad de corriente de 0,1 A g −1 durante 100 ciclos y con una densidad de corriente de 1 A g −1 para los siguientes 100 ciclos. Los ánodos nanocompuestos pSi @ NC mostraron una capacidad de 1720 mA h g −1 después de 110 ciclos a una densidad de corriente de 0,1 A g −1 , correspondiente a una retención de capacidad del 79%. Además, los electrodos compuestos pSi @ NC entregaron una capacidad reversible de 1010 mA h g −1 a 1 A g −1 después de 110 ciclos subsiguientes, con una tasa de disminución de la capacidad del 0,2% por ciclo desde el ciclo 101 al 210. La Figura 4d muestra el rendimiento de velocidad del electrodo pSi @ NC. El electrodo pSi @ NC alcanzó capacidades de descarga de 2360, 1690, 1570, 1470, 1320 y 850 mA h g −1 a la densidad de corriente de 0,1, 0,3, 0,5, 1,0, 2,0 y 5,0 A g −1 , respectivamente. La capacidad de descarga podría recuperarse a aproximadamente 2160 mA h g −1 cuando la densidad de corriente volvió a 0,1 A g −1 , lo que demuestra que el ánodo compuesto pSi @ NC tenía una excelente reversibilidad electroquímica. En comparación, el polvo de silicio comercial (Fig. 4e) recubierto con el carbono conductor dopado con nitrógeno como ánodo alcanzó una alta capacidad de descarga inicial de 3230 mA h g −1 , pero sufrió un deterioro severo de la capacidad a 110 mA h g - 1 después de 100 ciclos a 0,1 A g −1 . Estos resultados sugirieron que la capa conductora de carbono dopado con nitrógeno y la estructura porosa en pSi @ NC podrían proporcionar las vías de transporte rápido de iones / electrones y mantener la estabilidad estructural, dotando así al ánodo compuesto pSi @ NC de un buen rendimiento de velocidad y una excelente reversibilidad [ 21, 39, 60]. Además, la oxidación de la superficie en pSi también podría contribuir a mejorar la eficiencia del ciclo de las baterías de iones de litio, lo que limitó la expansión de volumen de las partículas de silicio y evitó algunas reacciones secundarias según los estudios anteriores [58].
Conclusiones
En resumen, desarrollamos un nuevo método para preparar silicio nanoporoso con altos rendimientos basado en la reacción de siliciuro de magnesio (Mg 2 Si) en líquido iónico ácido. Cuando se recubren con la capa de carbono dopado con nitrógeno y se aplican como ánodo de una batería de iones de litio, los compuestos de silicio-carbono obtenidos exhibieron una alta capacidad reversible, estabilidad cíclica a largo plazo y una alta eficiencia colónica inicial. La capa de revestimiento de carbono dopado con N suministró las vías conductoras eficientes para el transporte rápido de iones de litio y la transferencia de electrones, lo que es beneficioso para mejorar las propiedades electroquímicas de las partículas de silicio. Dado que la condición de reacción es relativamente suave y el rendimiento de los productos es superior al 82%, este método de preparación podría extenderse a la producción en masa de materiales de ánodos de silicio.
Abreviaturas
- [Bmim] Cl:
-
Cloruro de 1-butil-3-metilimidazolio
- AlCl 3 :
-
Cloruro de aluminio
- CV:
-
Voltamperometría cíclica
- EDS:
-
Espectroscopía de dispersión de energía
- Mg 2 Si:
-
Siliciuro de magnesio
- pSi:
-
Nanopartículas de silicio poroso
- pSi @ NC:
-
Carbón dopado con nitrógeno recubierto de nanopartículas de silicio poroso
- PXRD:
-
Difracción de rayos X en polvo
- SEM:
-
Microscopía electrónica de barrido
- TEM:
-
Microscopía electrónica de transmisión
- TGA:
-
Análisis termogravimétrico
- XPS:
-
Espectroscopia de fotoelectrones de rayos X
Nanomateriales
- Compuesto híbrido Nanoestructurado de sílice / oro-celulosa-Amino-POSS mediante el proceso Sol-Gel y sus propiedades
- Síntesis fácil de nanopartículas de SiO2 @ C ancladas en MWNT como materiales de ánodo de alto rendimiento para baterías de iones de litio
- Síntesis y propiedades electroquímicas de materiales de cátodo LiNi0.5Mn1.5O4 con dopaje compuesto Cr3 + y F− para baterías de iones de litio
- Compuesto negro de acetileno / MoS2 de pocas capas como material de ánodo eficiente para baterías de iones de litio
- Preparación de micromateriales híbridos de MnO2 recubiertos de PPy y su rendimiento cíclico mejorado como ánodo para baterías de iones de litio
- Compuesto de grafeno / Si integrado fabricado por reducción térmica de magnesio como material anódico para baterías de iones de litio
- Síntesis fácil y respetuosa con el medio ambiente de nanocables de Co3O4 y su prometedora aplicación con grafeno en baterías de iones de litio
- Síntesis sonoquímica de un solo paso y propiedades fotocatalíticas fáciles de compuestos de puntos cuánticos de grafeno / Ag3PO4
- Un ánodo de película de Fe2O3 nanocristalino preparado por deposición de láser pulsado para baterías de iones de litio
- Síntesis rápida de nanocristales de Pt y materiales de Pt / La2O3 microporosos mediante levitación acústica
- Síntesis y rendimiento del supercondensador de compuestos de carbono mesoporoso ordenados con polianilina / dopado con nitrógeno