


Identificación de una vía de disociación de excitones intramolecular específica de puente en polímeros conjugados alternados de donante-aceptor
Resumen
La disociación del excitón intramolecular es fundamental para las generaciones de portadores de carga móviles de alta eficiencia en las células solares orgánicas. Sin embargo, a pesar de mucha atención, los efectos de los puentes π sobre la dinámica de disociación del excitón en los polímeros conjugados alternativos donante-π-aceptor (D-π-A) siguen sin estar claros. Aquí, utilizando una combinación de espectroscopia de absorción transitoria (TA) resuelta en el tiempo de femtosegundos y espectroscopia de estado estacionario, rastreamos la dinámica de relajación intramolecular ultrarrápida del excitón en tres polímeros conjugados alternos D-π-A que fueron sintetizados por el grupo de Qin y llamados HSD-A , HSD-B, HSD-C. Se encuentra que la adición de unidades de tiofeno como puentes π conducirá al desplazamiento hacia el rojo del espectro de absorción en estado estacionario. Es importante destacar que revelamos la existencia de una nueva vía de disociación del excitón intramolecular mediada por un estado de transferencia de carga específica de puente (CT ') con el pico de huella dactilar TA a 1200 nm en HSD-B y HSD-C con puente π. Este estado CT 'da como resultado velocidades de captura de electrones más altas para HSD-B y HSD-C en comparación con HSD-A. Dependiendo de la proporción de estado CT ′ y la recombinación no genética son pasos importantes para comprender las eficiencias de conversión de alta potencia en HSD-B que en HSD-C. Proponemos que esta vía de disociación de excitones específica del puente juega un papel importante en la disociación de excitones intramolecular ultrarrápida de materiales fotovoltaicos orgánicos D-π-A alternando polímeros conjugados.
Introducción
Los dispositivos fotovoltaicos orgánicos (OPV) que utilizan energía solar para satisfacer la creciente demanda mundial de energía podrían considerarse como uno de los sustitutos más importantes de la producción de fuentes de energía limpias y renovables [1, 2, 3, 4]. Los polímeros conjugados alternados donante-aceptor, en los que bloques conjugados de diferente afinidad electrónica están dispuestos alternativamente a lo largo de la cadena principal de los polímeros, son un excelente material electrónico orgánico. El hecho de que muestre una eficiencia de conversión de energía (PCE) relativamente alta se debe en parte a las brechas ópticas más bajas que permiten una recolección más eficiente de fotones solares en el rango del infrarrojo cercano (NIR) [5]. En consecuencia, los dispositivos OPV que consisten en polímeros conjugados alternados de donante-aceptor podrían convertirse en alternativas económicamente viables a las células solares basadas en Si [6,7,8,9].
Las últimas investigaciones de varios grupos han demostrado que la eficacia de la OPV se puede mejorar mediante el uso de copolímeros alternos conjugados con banda prohibida baja (como se observa en las familias PCDTBT, PBDTTT y PTB) [10,11,12,13]. Una diferencia importante entre estos polímeros es que la transición del excitón al nivel de energía más bajo exhibe características de transferencia de carga parcial. Se considera que el estado de transferencia de carga intramolecular promueve la separación de carga final en la heterounión [14, 15, 16, 17, 18, 19]. Por tanto, es razonable esperar que las propiedades de los polímeros donantes de banda prohibida baja estén estrechamente relacionadas con el rendimiento de los dispositivos. Sin embargo, la conexión entre el rendimiento de los dispositivos y las propiedades inherentes de los polímeros aún es imprecisa. Especialmente, la división ultrarrápida del excitón y la dinámica del portador de los polímeros donantes de banda prohibida baja no están directamente relacionadas con el PCE de los dispositivos. Por un lado, una masa de curso paralelo y secuencial en escala temporal ultrarrápida y lenta solo se encuentra en condiciones relacionadas con el equipo. Por otro lado, sólo se podría considerar que la división del excitón en la interfaz de la heterounión en masa donante-aceptor (BHJ) condiciona el PCE del dispositivo [5]. Por lo tanto, es necesario estudiar la dinámica del excitón y del portador de los polímeros donantes de banda prohibida baja para optimizar el PCE de los dispositivos fotovoltaicos orgánicos D-A.
Recientemente, el grupo de Qin sintetizó una serie de polímeros conjugados alternos D-π-A [20, 21, 22]. Por ejemplo, los copolímeros HSD consisten en carbazol con enlaces 2,7 como unidad donante y 5,6-bis (octiloxi) benzo [c] [1,2,5] tiadiazol como unidad aceptora, mientras que tienen diferentes puentes π. El bloque donante y el bloque aceptor se polimerizan directamente para preparar el material fotovoltaico como HSD-A. De manera diferente, una unidad de tiofeno actúa como el bloque donante de conexión del puente π y el bloque aceptor se denota como HSD-B, así como el bloque donante y el bloque aceptor están conectados por dos unidades de tiofeno se denota como HSD-C. Descubrieron que los puentes π en los copolímeros tienen un efecto significativo sobre las propiedades de los copolímeros HSD. Los diferentes puentes π afectan críticamente la deslocalización electrónica de la cadena principal del polímero conjugado, la morfología de la película y las propiedades ópticas, electroquímicas, de transporte de carga y fotovoltaicas de los copolímeros HSD [23]. Uso de copolímeros HSD como donador de electrones y PC 71 BM como aceptor de electrones para preparar dispositivos fotovoltaicos orgánicos, se encuentra que los dispositivos preparados con polímeros HSD con diferentes puentes π tienen diferentes PCE. Los dispositivos OPV con HSD-A:PC71BM como capa activa demostraron que el PCE es bajo; HSD-B:PC71BM presenta un PCE del 5,4%; HSD-C:PC71BM muestra un PCE de 2,15% [20, 21]. Estas evidencias indican que los copolímeros donantes tienen un efecto sobre el PCE de las células solares poliméricas, pero la correlación entre el rendimiento de los dispositivos y las características inherentes de los copolímeros donantes, como la estructura, la energía y la dinámica del portador móvil, aún no se distingue. Los principales procesos de relajación que siguen a la absorción de luz son importantes para determinar el rendimiento de los dispositivos fotovoltaicos. Por lo tanto, es imperativo comprender la dinámica del excitón y del portador de los copolímeros HSD mediante el seguimiento de los excitones.
El rápido desarrollo de la tecnología láser ultracorta hace posible monitorear y rastrear la formación y ruptura de enlaces químicos en moléculas y varios procesos dinámicos dentro y entre moléculas con resolución de tiempo de femtosegundos y alta precisión espacial. Este trabajo aclara los procesos de disociación de excitones y relajación ultrarrápida de los copolímeros HSD utilizando una combinación de espectroscopía de absorción en estado estacionario y absorción transitoria de femtosegundos resuelta en el tiempo. Las bandas de espectros características se midieron y analizaron en detalle, revelando un mecanismo de relajación ultrarrápido para la dinámica de disociación de excitones. Nuestros resultados dan una mejor idea de las propiedades físicas de los copolímeros HSD y proporcionan una base experimental para mejorar el PCE de las células solares poliméricas.
Materiales y métodos experimentales
Materiales
HSD-A, HSD-B, HSD-C fueron proporcionados por el grupo de Qin, y la síntesis y caracterización de estos co-oligómeros se mostró en la literatura [20, 21]. Las estructuras moleculares de estos co-oligómeros se muestran en la Fig. 1a. La solución utilizada para preparar estos co-oligómeros fue o-diclorobenceno, con una concentración de aproximadamente 0,1 mg / ml. Esta concentración no solo puede garantizar que se pueda medir una buena señal resuelta en el tiempo, sino que también puede garantizar que el cromóforo esté completamente separado, de modo que el estado excitado no se apague con la intensidad de excitación utilizada [24].
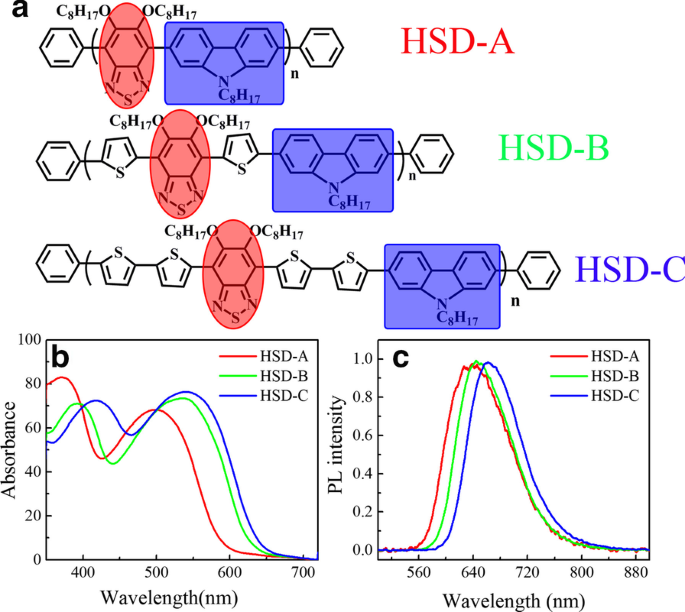
Estructuras moleculares de tres polímeros ( a ) en este trabajo. Sombreado de azul y sombreado de rojo indican las partes donante y aceptora, respectivamente. Espectros de absorción en estado estacionario ( b ) y espectros de fotoluminiscencia en estado estacionario ( c ) de tres muestras, HSD-A (rojo), HSD-B (verde), HSD-C (azul), medido en o-diclorobenceno
Medidas espectroscópicas
La espectroscopia de absorción en estado estacionario se midió con un espectrofotómetro de doble haz (Cary-5000, Agilent), y la espectroscopia de fluorescencia en estado estacionario se midió con un espectrómetro de fibra óptica (USB-4000, Ocean Optics).
La espectroscopía de absorción transitoria resuelta en el tiempo de femtosegundos se midió mediante láser de femtosegundos (Coherent), amplificador paramétrico óptico (OPA, TOPAS) y espectrómetro de absorción transitoria (fuego Helios). El láser de femtosegundos generado por el láser de femtosegundos se divide en dos recorridos a través de un divisor de haz (1:1), uno de los cuales ingresa al TOPAS y genera pulsos de bombeo con diferentes longitudes de onda; el otro haz pasa a través de un divisor de haz (2:98) nuevamente, y la pequeña parte del láser proyectado ingresa al espectrómetro de absorción transitoria Helios para generar pulsos de sonda continuos de luz blanca (WLC) (420–780 nm, 820–1600 nm ).
Resultados y discusión
La Figura 1a muestra la fórmula estructural de los polímeros conjugados HSD usados en este trabajo, los restos donantes están marcados con un recuadro azul y los restos aceptores están resaltados con un círculo rojo. La unidad de tiofeno actúa como puente específico entre el donante y el aceptor, evitando secuencialmente la repulsión estérica entre el donante y las unidades aceptoras individuales. Como se informó anteriormente, también puede lograr una separación de carga a larga distancia entre el donante y el aceptor, asegurando así un estado de transferencia de carga de larga duración [8]. La Figura 1b muestra los espectros de absorción en estado estacionario de los tres polímeros, y los espectros de absorción de los tres polímeros con diferentes puentes π presentan formas similares, caracterizadas por dos bandas de absorción distintas. El perfil típico de dos picos también se ha informado en otros polímeros conjugados, que es una característica única de los polímeros conjugados D-A [25,26,27,28,29,30]. Los picos de absorción de HSD-A están alrededor de 370 y 490 nm, y los de HSD-B están alrededor de 390 y 530 nm, y los de HSD-C están alrededor de 420 y 540 nm. Estos dos picos de absorción se atribuyen a la transición π – π * con el pico de energía más bajo asociado con la transferencia de carga intracadena [31]. Las posiciones de los picos de absorción en estado estacionario se ven afectadas por el reemplazo de diferentes unidades de puentes π, lo que resulta en el corrimiento al rojo de los picos de absorción debido principalmente al efecto de deslocalización de electrones [32]. Hemos realizado cálculos químicos cuánticos en polímeros, y los orbitales moleculares de frontera de los polímeros HSD se han calculado y proporcionado en la Fig. 2a. El orbital molecular ocupado más alto (HOMO) y el orbital molecular desocupado más bajo (LUMO) de las tres muestras se muestran en la Fig. 2a, y las brechas de energía HOMO – LUMO (ΔE H – L ) se representan en la Fig. 2b. Puede verse claramente en la Fig. 2b que las brechas de energía HOMO – LUMO (ΔE H – L ) de HSD-A a HSD-B y HSD-C disminuyen gradualmente, lo que es coherente con el desplazamiento hacia el rojo en los espectros de HSD-A a HSD-B y HSD-C [33]. La Figura 1c presenta los espectros de fotoluminiscencia (PL) de las tres muestras en su solución. Los espectros PL de las tres muestras son igualmente sellables y consistentes con los espectros de absorción en estado estacionario. Es de destacar que sus picos se mueven hacia la onda larga con el aumento en los números de tiofeno. Los puentes π de los polímeros HSD pueden sintonizar el PCE de las células solares orgánicas fabricadas con la mezcla de polímero HSD y PC71BM, el PCE de los dispositivos que usamos en este estudio se enumera en el siguiente orden:HSD-B> HSD-C> HSD- A [20, 21].
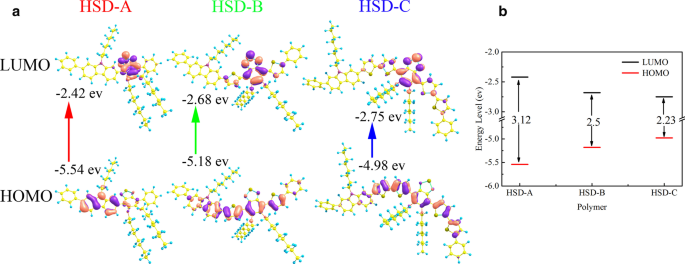
Orbitales moleculares de frontera de copolímeros HSD ( a ) calculado utilizando la función B3LYP-D3 con un conjunto básico de 6-311G ** y los niveles de energía HOMO y LUMO ( b )
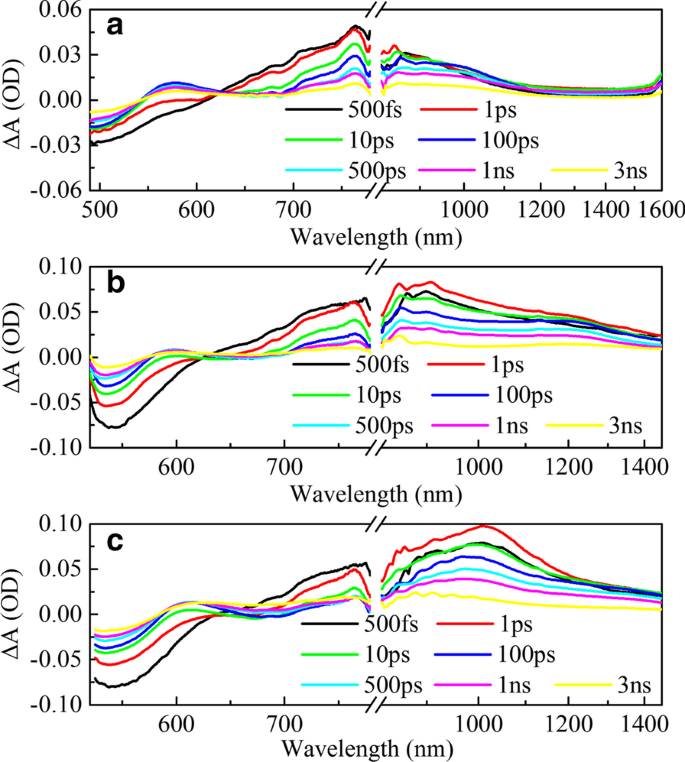
La espectroscopia de estado estacionario solo puede proporcionar una macrodescripción de los estados de transición electrónicos generales. Para investigar cómo los puentes π afectan el PCE de los dispositivos, realizamos mediciones de absorción transitoria de los tres polímeros HSD como se muestra en la Fig. 3. Los gráficos de los rangos visibles (VIS) de las tres muestras (Fig. 3a– c) son similares, mostrando tres características espectrales. La señal negativa (azul claro en el mapa) a aproximadamente 500 nm se asigna a la señal de blanqueo en estado fundamental (GSB), porque corresponde bien al segundo pico de absorción estable como se muestra en la Fig. 1b. Las tres muestras tienen dos señales de absorción positivas (rojo claro en el mapa) en el rango visible, y los picos de absorción están a 600 nm y 750 nm, respectivamente, lo que se considera absorción en estado excitado (ESA) [34]. En el rango de detección del infrarrojo cercano (NIR) (Fig. 3d-f), las tres muestras muestran diferencias obvias. HSD-A casi no tiene señal de absorción en el rango del infrarrojo cercano, pero HSD-B y HSD-C tienen una señal de absorción roja de área grande dentro del alcance de 800-1500 nm.
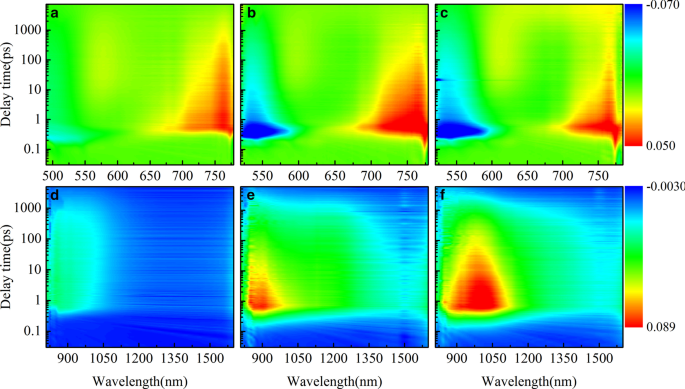
Espectros de absorción transitoria (TA) resueltos en el tiempo de femtosegundos a diferentes longitudes de onda de sonda de diferentes muestras. Mapas bidimensionales en función de la longitud de onda de la sonda (500-1600 nm) en la excitación a una longitud de onda de 470 nm del HSD-A ( a , d ); excitación a 500 nm de longitud de onda del HSD-B ( b , e ); excitación a 500 nm de longitud de onda del HSD-C ( c , f )
La Figura 4 muestra la evolución temporal de los espectros de absorción diferencial de los tres polímeros HSD en una escala de tiempo ultrarrápida. En el rango de 490-780 nm, la excitación a baja energía de fotones induce una amplia señal positiva en la región espectral de 700-780 nm que se eleva rápidamente con la señal negativa en la región espectral de 490-600 nm como GSB. Asignamos la amplia señal positiva de 700 a 780 nm al estado de excitón (EX) que se atribuye a la absorción de la siguiente manera. En primer lugar, su vida útil es coherente con la vida útil de otros excitones en los polímeros aislados en la literatura de 500 a 1000 ps [35] y es mucho más corta que la vida útil del estado de transferencia de carga (CT) y el estado de carga separada (CS) , que tiene una escala de tiempo superior a un par de nanosegundos. En segundo lugar, tiene una tendencia dinámica similar a GSB en los primeros cientos de picosegundos. Otra señal positiva (con un pico de aproximadamente 600 nm) aparece después de unos pocos picosegundos, que corresponde a la formación del estado de portadora móvil (MC). Dado que los espectros de aproximadamente 600 nm podrían atribuirse razonablemente al hecho de que la superposición de la absorción de GSB y MC en los experimentos de absorción transitoria. La señal negativa inicial a 600 nm se debe al hecho de que la señal de GSB es mucho más fuerte que la señal de absorción de MC. A medida que aumenta el tiempo de retardo, aparece la característica TA positiva cuando la absorción de MC es más fuerte que la del GSB. Además, la razón del hundimiento a 650 nm es que el estado excitado inestable vuelve al estado fundamental debido a la emisión estimulada (EE) y la consistencia espectral con la fluorescencia del estado estable. En los rangos de NIR, se puede ver que las señales de absorción de las tres muestras aumentaron dentro de 1 ps con un pico de aproximadamente 1 ps y luego muestran una tendencia a atenuarse. Curiosamente, la forma y la tendencia de atenuación de las señales de absorción de las tres muestras son diferentes. Para analizar estas diferencias con más detalle, realizamos un ajuste de picos en los espectros infrarrojos, y los resultados se muestran en la Fig. 5.
Espectros de diferencia asociados a la evolución (EADS) en VIS – NIR de HSD-A ( a ), HSD-B ( b ), HSD-C ( c )
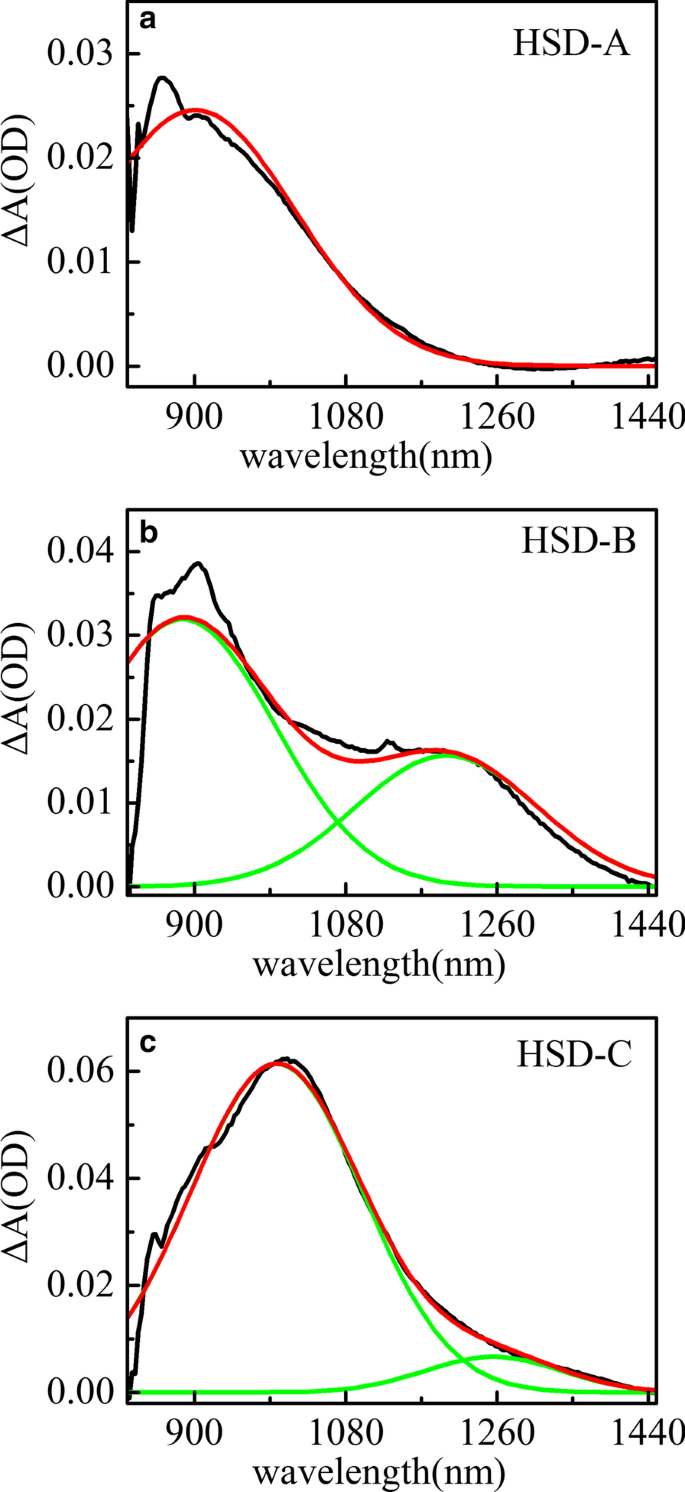
Pico espectral de absorción transitoria ajustado de HSD-A ( a ) y HSD-B ( b ) y HSD-C ( c ) en el tiempo de retardo de 2 ps en el rango de infrarrojos. La curva negra representa el espectro de absorción de las muestras a 2 ps, la curva roja es el espectro de absorción ajustado y la verde es la señal espectral identificada en el espectro
La Figura 5 muestra el ajuste de pico del espectro de absorción transitoria de HSD-A (a) y HSD-B (b) y HSD-C (c) en el tiempo de retardo de 2 ps en el rango de infrarrojos. En HSD-A, los espectros podrían ajustarse bien mediante análisis de un componente, mientras que en HSD-B y HSD-C, estos espectros podrían aproximarse al máximo mediante análisis de dos componentes diferentes. Esto significa que HSD-B y HSD-C tienen una señal de absorción más en el rango de infrarrojos que la dosis de HSD-A, que se debe a la adición del puente de tiofeno. La adición del puente de tiofeno amplía el rango de absorción de los polímeros en NIR, lo que permite que HSD-B y HSD-C tengan un nuevo pico de absorción cercano a los 1200 nm. Al principio, estas dos señales positivas tienen un tiempo de subida estrechamente relacionado con la atenuación de los picos EX como se muestra en la Fig. 6, que muestra que estas señales positivas son generadas directamente por los estados EX. Las señales positivas cercanas a 900 nm en las tres muestras podrían asignarse al estado de transferencia de carga intramolecular (CT). En este estado, los excitones se dividen en pares hueco-electrón, y los pares hueco-electrón todavía están lo suficientemente cerca como para generar la gravedad de Coulomb [36, 37]. Otras nuevas señales positivas cercanas a 1200 nm solo están presentes en HSD-B y HSD-C y también están acompañadas por la atenuación de EX en el período inicial, pero tienen una tendencia de atenuación diferente del estado CT en una ventana de tiempo prolongada. Este es un nuevo canal de disociación de excitones y lo consideramos el estado CT ′. Porque tiene características similares al estado CT, pero la dinámica de desintegración es diferente del estado CT.
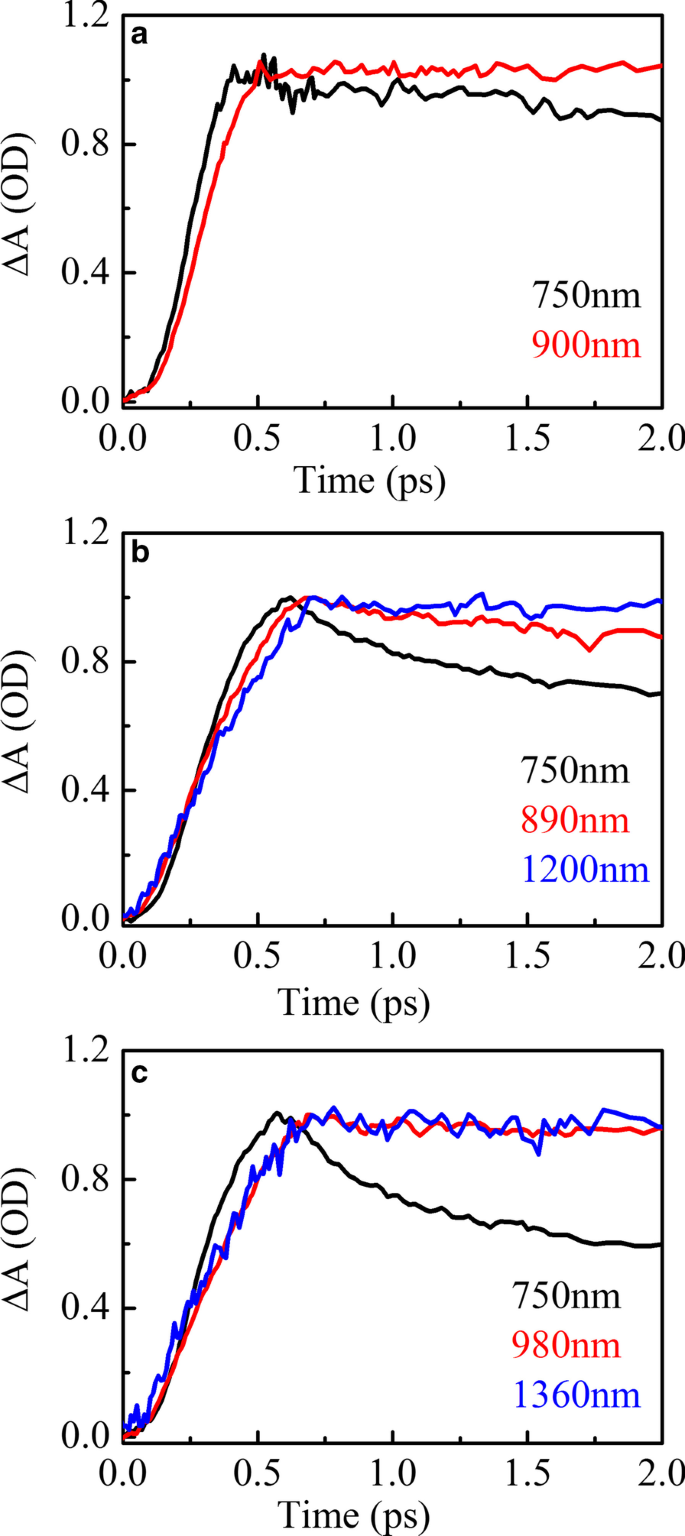
Absorción transitoria EX (negro) y CT (rojo) y cinética de CT ′ (azul) para HSD-A ( a ) y HSD-B ( b ) y HSD-C ( c ), exhibiendo la desintegración EX simultáneamente con el aumento en CT y CT ′
Como se muestra en la Fig.7, las curvas dinámicas del estado MC, estado EX, estado CT y CT ′ en HSD-A, HSD-B y HSD-C se extraen y ajustan, respectivamente, que representan la evolución en el tiempo de diferentes componentes. La fórmula de ajuste para las curvas dinámicas es ∆A (t) =a 1 exp (- t / τ 1 ) + A 2 exp (- t / τ 2 ) + ··· + a n exp (- t / τ n ), donde un 1 , un 2 ,… A n son amplitudes, τ 1 , τ 2 ,…, Τ n corresponden a constantes de tiempo [38, 39]. La Tabla 1 enumera los componentes de tiempo ajustados y las amplitudes relativas. A modo de comparación, las amplitudes máximas están normalizadas. Puede verse claramente a partir de los datos del ajuste del estado de MC (Fig. 7a, b) que la velocidad de generación de portadora de HSD-A es la más rápida. Su vida de formación es 6.43 ps, que es menor que 12.6 ps de HSD-B y 8.41 ps de HSD-C. Sin embargo, los portadores de HSD-A decaen rápidamente después de formarse, mientras que HSD-B y HSD-C tienen un proceso de aumento lento adicional con la constante de tiempo de 28,8 ps y 26,4 ps, respectivamente. Esto podría dificultar la captura de los portadores en HSD-A, lo que probablemente sea una de las razones del PCE más bajo de los dispositivos. En el estado EX (Fig. 7c, d), las tendencias de desintegración de las tres muestras son obviamente diferentes. HSD-A tiene una vida de desintegración significativamente más larga, por lo que la división del excitón es relativamente lenta. En HSD-B y HSD-C, hay tres tiempos de decaimiento en 1 ns, un femtosegundo (0,712 ps para HSD-B, 0,408 ps para HSD-C) y un picosegundo (18,4 ps para HSD-B, 7,96 ps para La vida útil de HSD-C) representa la transición del estado EX a otros estados. La vida útil más larga de cientos de picosegundos (735 ps para HSD-B, 627 ps para HSD-C) es del mismo orden de magnitud que la vida útil del excitón informada anteriormente del P3HT aislado. Por lo tanto, se puede considerar que \ (\ uptau _ {3} ^ {{{\ text {EX}}}} \) en el ajuste EX es más probable que sea la vida útil del excitón sin el proceso de transición [35, 40] . Sin embargo, en una ventana de tiempo prolongada, la recombinación de excitones ocurre en HSD-C. La cinética de CT (Fig. 7e, f) de estas tres muestras se ajusta mejor mediante tres componentes de vida útil. La corta constante de tiempo creciente, \ (\ uptau _ {1} ^ {{{\ text {CT}}}} <1 \) ps, está estrechamente relacionada con el tiempo de vida de decaimiento concurrente del estado EX, lo que significa la transición de Estado EX al estado CT. De manera similar, también existe una buena relevancia entre la vida útil de la desintegración del estado CT \ (\ uptau _ {2} ^ {{{\ text {CT}}}} \) y la vida útil creciente del estado portador \ (\ uptau _ {2} ^ {{{\ text {MC}}}} \), que indica la transición del estado CT al estado MC. Al comparar la vida útil de la caída del estado de CT de las tres muestras, se puede encontrar que la vida útil de la caída del estado de CT de HSD-B es significativamente más corta que la de HSD-A y HSD-C, lo que indica que HSD-B tiene una pérdida de estado de CT más rápida. calificar. El estado CT ′ (Fig. 7g, h) de HSD-B y HSD-C exhibe diferentes curvas dinámicas del estado CT en el intervalo de 50 ps. Tiene una vida útil de transición más larga \ (\ uptau _ {2} ^ {{{\ text {CT}} ^ {{\ prime}}}} \), que está en buena correlación con \ (\ uptau _ {3} ^ {{{\ text {MC}}}} \), que representa la transición del estado CT ′ al estado MC.
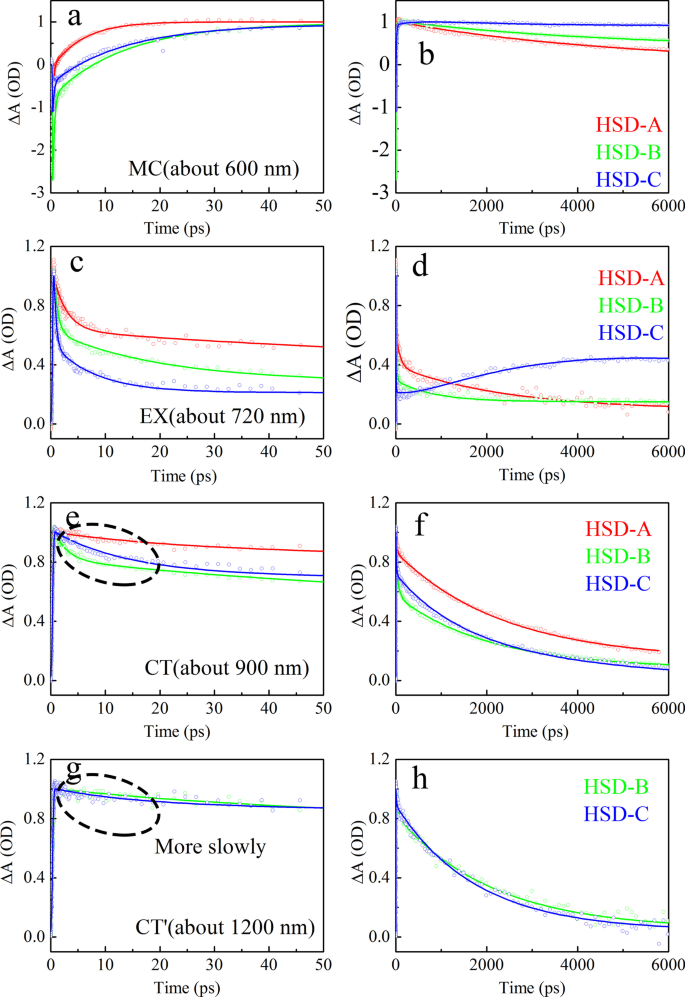
Ajuste cinético de todas las características espectrales transitorias de tres muestras. La figura muestra la cinética de 50 ps ( a , c , e ) y 5000 ps ( b , d , f ). Los ajustes son para MC ( a , b ), EX ( c , d ), CT ( e , f ), CT ′ ( g , h ) características del espectro
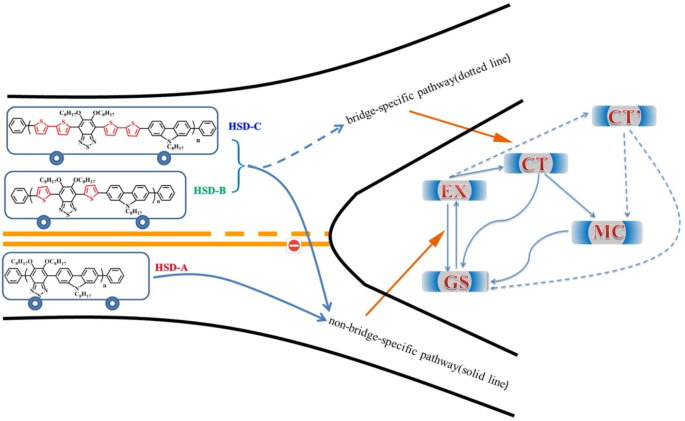
En la figura 8 se propone un esquema del diagrama de energía simplificado para las vías de relajación de los excitones. La diferencia en la conformación local en los polímeros conduce a una variación de energía en diferentes estados. Por lo tanto, estos estados muestran diferentes características de AT. En los polímeros HSD, el estado MC, el estado EX, el estado CT y el estado CT 'se proponen inevitablemente para dar una explicación más razonable al mecanismo de relajación del excitón. Los excitones generados después de la excitación de la luz se dividirán rápidamente en el estado CT y el estado CT ′. En estos estados, los excitones se dividen en pares hueco-electrón y todavía están lo suficientemente cerca como para experimentar una atracción de Coulombic. Con el retardo de tiempo, el agujero de electrones del estado CT y el estado CT ′ continuarán dividiéndose en estados MC más estables. Es importante destacar que encontramos que el estado CT 'solo existe en HSD-B y HSD-C con puentes de tiofeno, lo que agrega un nuevo canal de división de excitones a los polímeros HSD. Esto dará como resultado tasas de captura de electrones más altas para HSD-B y HSD-C, lo que es consistente con el PCE más alto de HSD-B y HSD-C en comparación con HSD-A. Mientras tanto, el hecho de que el PCE de HSD-C sea más bajo que el de HSD-B podría explicarse razonablemente de la siguiente manera:(a) la adición de dos tiofenos como puente π podría aumentar la separación espacial entre D y A, dando como resultado un recombinación en HSD-C. (b) Como se ve en la Fig. 5, la proporción de estado CT 'de HSD-B es significativamente mayor que la de HSD-C, lo que determina el PCE completo de HSD-B. Por lo tanto, el estado CT ′ en los polímeros HSD es fundamental para que una mayor capacidad de captura de electrones resulte en una mayor PCE de los dispositivos poliméricos HSD.
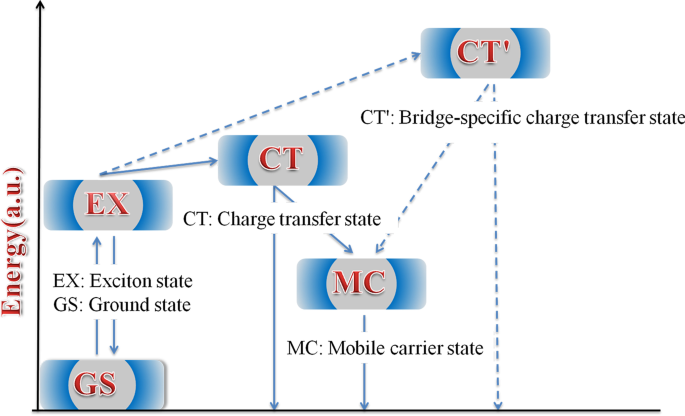
Ilustración esquemática de los estados y vías en la dinámica de división de excitones de los polímeros HSD en solución
Conclusiones
En general, utilizamos una combinación de espectroscopía de absorción transitoria y de estado estacionario para estudiar el impacto de los puentes π en los copolímeros HSD. Se encontró que la adición de la unidad de tiofeno como puente π conducirá al desplazamiento hacia el rojo del espectro de absorción en estado estacionario. Mientras tanto, los datos de absorción transitoria indican que HSD-B y HSD-C con unidad de tiofeno como puente π tenían un estado CT ′ adicional con el pico de huella dactilar TA a 1200 nm, lo que agrega un nuevo canal de disociación de excitones para polímeros HSD. La presencia del estado CT 'hace que el polímero sea ventajoso para la conversión fotoeléctrica. Entre los tres polímeros HSD estudiados en este trabajo, solo HSD-B y HSD-C que contienen puentes π tienen estados CT ′. Por tanto, creemos que la existencia de puentes π promueve la generación de estados CT ′. Sin embargo, el puente π no puede ser demasiado largo. Por ejemplo, el polímero HSD-C con dos tiofenos como puente π da como resultado una recombinación no geminada debido a la existencia del puente π demasiado largo y afecta la proporción de estados CT ′. Además, también aclaramos las vías de relajación del excitón analizando el ajuste dinámico de todas las características espectrales transitorias. Estos hallazgos proporcionan información fotofísica importante para mejorar la eficiencia de conversión de energía de los polímeros conjugados y un mayor desarrollo de las células solares orgánicas.
Disponibilidad de datos y materiales
Los conjuntos de datos utilizados y analizados en el estudio actual pueden obtenerse de los autores correspondientes previa solicitud razonable.
Abreviaturas
- D-π-A:
-
Donante-π-aceptor
- TA:
-
Absorción transitoria
- CT ′:
-
Estado de transferencia de carga específico del puente
- OPV:
-
Fotovoltaica orgánica
- PCE:
-
Eficiencias de conversión de energía
- NIR:
-
Infrarrojo cercano
- BHJ:
-
Heterounión masiva
- TOPAS:
-
Amplificador paramétrico óptico
- WLC:
-
Continuo de luz blanca
- PL:
-
Fotoluminiscencia
- VIS:
-
Visible
- GSB:
-
Blanqueamiento en estado fundamental
- ESA:
-
Absorción de estado emocionado
- EADS:
-
Espectros de diferencia asociados a la evolución
- EX:
-
Estado de excitación
- CT:
-
Estado de transferencia de carga
- CS:
-
Estado de carga separada
- MC:
-
Estado del operador de telefonía móvil
- SE:
-
Emisión estimulada
Nanomateriales
- ¿Qué es la corriente alterna (CA)?
- Códigos de identificación de resina
- Lanzamiento de polímeros estirénicos renovables
- Detección de excitación espacialmente localizada en superredes de puntos cuánticos autoorganizadas InAs / InGaAs:una forma de mejorar la eficiencia fotovoltaica
- Fabricación, caracterización y citotoxicidad de nanopartículas de carbonato de calcio derivadas de concha de oro-berberecho conjugado de forma esférica para aplicaciones biomédicas
- Identificación de macromoléculas características de genotipos de Escherichia coli mediante microscopio de fuerza atómica Mapeo mecánico a nanoescala
- Preparación de nanogeles poliméricos termorresistentes de ácido diacrilato-metacrílico de oligo (etilenglicol) y caracterización de sus propiedades
- Catalizadores a base de platino en varios soportes de carbono y polímeros conductores para aplicaciones de celdas de combustible de metanol directo:una revisión
- Polímeros piezoeléctricos
- Enfoque químico para una electrónica blanda más robusta
- Mecanizado CNC de polímeros