


Baja degradación por tunelización de uniones de una sola molécula de alcanos terminados en yodo
Resumen
Un tema clave para el desarrollo de dispositivos electrónicos moleculares es comprender el transporte de electrones de las uniones de una sola molécula. En este trabajo, exploramos el transporte de electrones de uniones moleculares simples de alcanos terminados en yodo utilizando el enfoque de unión de ruptura basado en microscopio de efecto túnel de barrido. El resultado muestra que la conductancia disminuye exponencialmente con el aumento de la longitud molecular con una constante de desintegración β N =0.5 por –CH 2 (o 4 nm −1 ). Es importante destacar que la desintegración tunelizada de esas uniones moleculares es mucho menor que la de las moléculas de alcano con tiol, amina y ácido carboxílico como grupos de anclaje e incluso comparable a la de las moléculas de oligofenilo conjugado. La baja desintegración por efecto túnel se atribuye a la pequeña altura de la barrera entre la molécula de alcano terminada en yodo y el Au, lo que está bien respaldado por los cálculos de DFT. El trabajo sugiere que el grupo de anclaje puede ajustar eficazmente la desintegración del túnel, lo que puede guiar la fabricación de cables moleculares.
Antecedentes
Comprender el transporte de electrones de las uniones de una sola molécula es crucial para el desarrollo de dispositivos electrónicos moleculares [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16 ]. El modelo de tunelización no resonante se ha utilizado a menudo para describir el proceso de transporte de electrones a través de moléculas pequeñas, donde la conductancia de contacto, la longitud molecular y la constante de desintegración de tunelización son los parámetros principales [17, 18]. En la mayoría de los sistemas moleculares, la constante de desintegración está muy relacionada con las propiedades electrónicas de la estructura orgánica. Por ejemplo, los sistemas moleculares conjugados tienen baja desintegración de túnel, a diferencia de los no conjugados [17, 19]. Dado que la desintegración del túnel se decide por la altura de la barrera entre el nivel de Fermi del electrodo y el orbital molecular desocupado más bajo (LUMO) o el orbital molecular ocupado más alto (HOMO) de las uniones moleculares [17, 20], es posible ajustar el nivel de energía molecular hacia el nivel de Fermi para lograr la baja desintegración [21,22,23,24].
En las uniones de una sola molécula, el grupo de anclaje juega un papel importante en el control del acoplamiento electrónico entre las cadenas principales orgánicas con los electrodos [21, 23, 24, 25]. Una serie de medidas de conductancia para las moléculas basadas en alcanos ha mostrado un efecto significativo de diferentes grupos de anclaje sobre la geometría de unión, las probabilidades de formación de uniones, la conductancia de contacto e incluso el canal de conductancia (a través de LUMO o HOMO) de las uniones moleculares [21,22, 23,24,25]. Dado que el grupo de anclaje puede regular los orbitales de la frontera en la unión molecular, el grupo de anclaje también puede ajustar la desintegración de túnel de la molécula [24]. Sin embargo, se han realizado estudios limitados en esta área.
En este documento, informamos sobre el transporte de electrones de las moléculas de alcano terminadas con el grupo yodo mediante el uso de microscopía de efecto túnel de barrido (STM-BJ) (Fig. 1) [26, 27]. Las mediciones de conductancia molecular única muestran que la conductancia disminuye exponencialmente con el aumento de las longitudes moleculares y la constante de desintegración de las moléculas de alcano con grupo yodo es mucho más baja que la de los análogos con otros grupos de anclaje. Las diferentes constantes de desintegración de túnel para moléculas de alcano con grupos de anclaje variados se explican por la altura de la barrera entre la molécula y el electrodo.
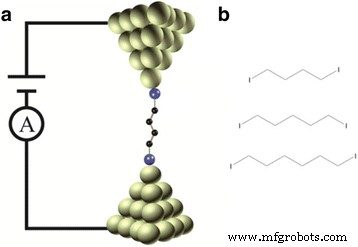
Diagrama esquemático de la unión de rotura de microscopía de efecto túnel de barrido (STM-BJ) y estructuras moleculares. un Esquema del STM-BJ con unión molecular. b Estructuras moleculares de moléculas de yodo alcano
Métodos
El 1,4-butanodiiodo, el 1,5-pentanodiiodo y el 1,6-hexanodiiodo se adquirieron de Alfa Aesar. Todas las soluciones se prepararon con etanol. Se utilizó Au (111) como sustrato, mientras que como puntas se utilizaron puntas de Au cortadas mecánicamente. Antes de cada experimento, el Au (111) se pulió electroquímicamente y se recoció cuidadosamente en una llama de butano y luego se secó con nitrógeno.
El sustrato de Au (111) se sumergió en una solución de etanol recién preparada que contenía moléculas diana 0,1 mM durante 10 min. La medición de la conductancia se llevó a cabo en el Nanoscopio IIIa STM modificado (Veeco, EE. UU.) Utilizando el método STM-BJ a temperatura ambiente [28,29,30], que simplemente midió la conductancia de las uniones de una sola molécula formadas por movimientos repetidos la punta dentro y fuera del sustrato a una velocidad constante. Durante el proceso, las moléculas podrían anclarse entre los dos electrodos metálicos y formar uniones moleculares únicas. Se recopilaron miles de estas curvas para el análisis estadístico. Todos los experimentos se realizaron con un voltaje de polarización fijo de 100 mV. Dado que las moléculas con yodo como grupo de anclaje son un material fotosensible, el experimento se llevó a cabo bajo sombra.
Resultados y discusión
Medición de la conductancia de uniones moleculares simples de alcanos terminados en yodo
Las medidas de conductancia se llevaron a cabo primero en Au (111) con monocapa de 1,4-butanodiiodo por STM-BJ. La Figura 2a muestra los trazos de conductancia típicos que exhiben la característica escalonada. Los rastros de conductancia muestran una meseta en 1 G 0 , lo que indica la formación de un contacto atómico estable de Au. Meseta con un valor de conductancia de 10 −3,6 G 0 (19.47 ns) también se encuentra además del 1 G 0 , debido a la formación de uniones moleculares. También se podría obtener un histograma de conductancia tratando con logaritmo y agrupando el valor de conductancia de más de 3000 trazas de conductancia, y luego, la intensidad del histograma de conductancia se normalizó por el número de trazas utilizadas y muestra un pico de conductancia en 10 - 3.6 G 0 (19,44 ns) (figura 2b). Aquellos muestran que el grupo yodo puede servir como un grupo de anclaje eficaz que forma una unión molecular. Sin embargo, este valor es menor que el valor de conductancia molecular única de 1,4-butanodiamina con amina como grupo de anclaje, que puede deberse a una interacción débil entre el yodo y el electrodo de Au [31].
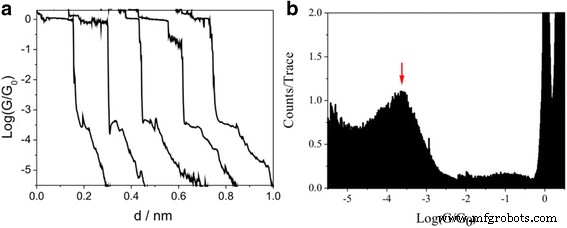
Conductancia molecular única de las uniones Au-1,4-butanodiiodo-Au. un Curvas de conductancia típicas de las uniones Au-1,4-butanodiiodo-Au medidas con un sesgo de 100 mV. b Histograma de conductancia en escala logarítmica de uniones 1,4-butanodiiodo con contactos de Au
En comparación con el 1,4-diyodobutano, picos pronunciados a 10 −3,8 G 0 (12,28 ns) y 10 −4,0 G 0 (7,75 ns) se encuentran para 1,5-pentanediiodo y 1,6-hexanodiiodo, respectivamente (Fig. 3). Los valores de conductancia disminuyen con el aumento de la longitud de la molécula. Mientras tanto, los valores de conductancia de 1,5-pentanodiiodo y 1,6-hexanodiiodo son menores que los de 1,5-pentanodiamina y 1,6-hexanodiamina, respectivamente [31], lo que puede deberse a la diferente interacción en alcano- uniones moleculares basadas entre el yodo y los grupos de anclaje de amina que se unen a los electrodos de Au [32].
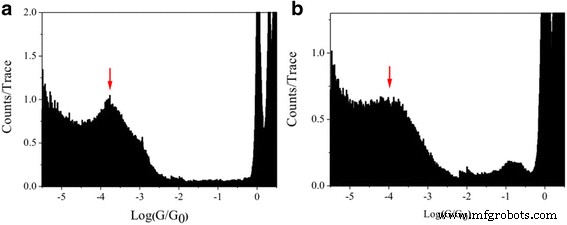
Conductancia molecular única de 1,5-pentanodiiodo y 1,6-hexanodiiodo con electrodo de Au. Histograma de conductancia a escala logarítmica de uniones moleculares individuales con a 1,5-pentanediiodo y b 1,6-hexanodiiodo
Los histogramas de conductancia bidimensional también se construyeron para esas uniones moleculares (archivo adicional 1:Figura S1) y dan valores de conductancia similares de histogramas unidimensionales. Normalmente, la distancia de ruptura de las uniones moleculares aumenta con el aumento de la longitud molecular. También analizamos la distancia desde el valor de conductancia de 10 −5.0 G 0 a 10 −0,3 G 0 como se muestra en la Fig. 4, y se encuentran distancias de ruptura de 0,1, 0,2 y 0,3 nm para 1,4-butanodiiodo, 1,5-pentanodiiodo y 1,6-hexanodiiodo, respectivamente. Aquí, las distancias de ruptura se obtienen del pico máximo del histograma de distancia de ruptura [33]. Se informó que hay una distancia de retroceso de 0.5 nm para Au después de la ruptura del contacto Au-Au [34, 35]; por tanto, las distancias absolutas para esas uniones moleculares entre electrodos podrían ser 0,6, 0,7 y 0,8 nm que se encuentran para 1,4-butanodiiodo, 1,5-pentanodiiodo y 1,6-hexanodiiodo, respectivamente. Esas distancias son comparables a la longitud de las moléculas. Eder y col. informó que la adsorción de la monocapa de 1,3,5-tri (4-yodofenil) -benceno sobre Au (111) puede causar deshalogenación parcial [36]; sin embargo, se puede encontrar un valor de conductancia muy mayor para esas uniones moleculares de contacto covalente de Au-C para moléculas con cuatro (alrededor de 10 −1 G 0 ) y seis (mayor que 10 −2 G 0 ) –CH 2 - unidades [37]. Por lo tanto, proponemos que las moléculas investigadas actualmente entren en contacto con el Au a través del contacto Au-I.
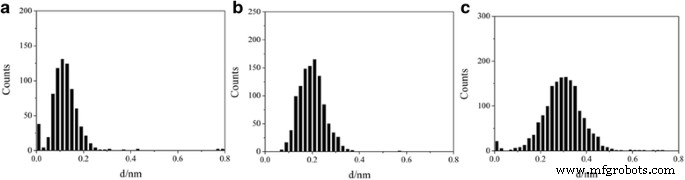
Rompiendo distancias para alcanos terminados en yodo. Rompiendo distancias de a 1,4-butanodiiodo, b 1,5-pentanediiodo y c 1,6-hexanodiyodo obtenido a partir de curvas de conductancia entre 10 −5.0 G 0 y 10 −0,3 G 0
Constante de desintegración de túneles de uniones moleculares individuales de alcanos terminados en yodo
Bajo el sesgo actual, la conductancia de esas moléculas se puede expresar como G = G c exp (- β N N ). Aquí, G es la conductancia de la molécula y G c es la conductancia de contacto y está determinada por la interacción entre el grupo de anclaje y el electrodo. N es el número de metileno en la molécula y β N es la constante de desintegración tunelizada, que refleja la eficiencia de acoplamiento del transporte de electrones entre la molécula y el electrodo. Como se muestra en la Fig. 5, graficamos una escala de conductancia logarítmica natural frente al número de metileno; constante de desintegración de túnel β N de 0,5 por –CH 2 se determina a partir de la pendiente del ajuste lineal. Esta desintegración por efecto túnel es muy baja en moléculas basadas en alcanos. Para las moléculas basadas en alcanos, β N generalmente se encuentra alrededor de 1.0 por –CH 2 para tiol (SH) [23, 38], mientras que alrededor de 0,9 y 0,8 por –CH 2 se determinan para amina (NH 2 ) [23, 31] y ácido carboxílico (COOH), respectivamente [39]. Por lo tanto, la desintegración de túnel con yodo muestra el valor más bajo entre los grupos de anclaje con una tendencia β N (tiol)> β N (amina)> β N (ácido carboxílico)> β N (yodo), que puede deberse a la diferencia en la alineación de los niveles de energía molecular al nivel de Fermi del electrodo de Au [23, 31]. La caída de tunelización de 0.5 por –CH 2 también se puede convertir a 4 nm −1 , que es comparable a los oligofenilos con 3,5–5 nm −1 [40, 41].
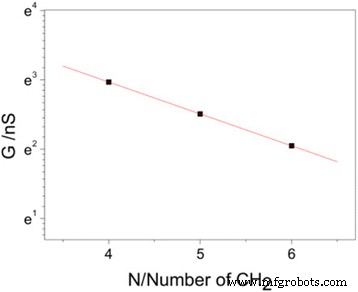
Conductancia de molécula única frente a longitud molecular para alcanos terminados en yodo. Gráficos logarítmicos de conductancia de una sola molécula frente a longitud molecular para alcanos terminados en yodo
El β N para las uniones metal-molécula-metal se puede describir simplemente mediante la siguiente ecuación [17, 20, 38],
$$ {\ beta} _N \ \ alpha \ \ sqrt [2] {\ frac {2 m \ varPhi} {h ^ 2}} $$
donde m es la masa efectiva del electrón y 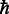 es la constante de Planck reducida. Φ representa la altura de la barrera, que se decide por la brecha de energía entre el nivel de Fermi y los niveles de energía molecular en la unión. Obviamente, el β N El valor es proporcional a la raíz cuadrada de la altura de la barrera. Por lo tanto, podemos proponer que las moléculas de alcanos terminados en yodo tienen pequeñas Φ con el electrodo de Au.
es la constante de Planck reducida. Φ representa la altura de la barrera, que se decide por la brecha de energía entre el nivel de Fermi y los niveles de energía molecular en la unión. Obviamente, el β N El valor es proporcional a la raíz cuadrada de la altura de la barrera. Por lo tanto, podemos proponer que las moléculas de alcanos terminados en yodo tienen pequeñas Φ con el electrodo de Au.
Altura de barrera de uniones moleculares individuales con diferentes grupos de anclaje
Tomando el - (CH 2 ) 6 - como columna vertebral, realizamos los cálculos aproximados (ver detalles computacionales en el archivo adicional 1) para investigar los orbitales moleculares de frontera de complejos con cuatro átomos de Au en ambos extremos, incluido 1,6-hexanoditiol (C6DT), 1,6- hexanodiamineb (C6DA), ácido 1,6-hexanodicarboxílico (C6DC) y 1,6-hexanodiyodo (C6DI). Como se muestra en la Tabla 1, HOMO y LUMO son - 6.18 y - 1.99 eV, respectivamente, para C6DT, mientras que HOMO (6.02 eV) y LUMO (- 1.85 eV) se encuentran para C6DA. Mientras tanto, los niveles de energía HOMO y LUMO se calculan para C6DC (-6,33 y -2,58 eV) y C6DI (-6,22 y -2,61 eV).
Para el nivel de Fermi del electrodo de Au, debemos considerar la influencia de la adsorción de moléculas. En la condición de vacío, el Au limpio da una función de trabajo de 5,1 eV [42]; mientras tanto, este valor puede modificarse obviamente mediante la adsorción de moléculas. Kim y col. [43] y Yuan et al. [44] han descubierto que la función de trabajo del Au es de alrededor de 4,2 eV (4,0–4,4 eV) sobre las monocapas autoensambladas adsorbidas (SAM) medidas por el espectrómetro de fotoelectrones ultravioleta (UPS). Low et al. también investigó el transporte de electrones de moléculas basadas en tiofeno de TOTOT (LUMO - 3.3 eV, HOMO - 5.2 eV) y TTO p TT (LUMO - 3.6 eV, HOMO - 5.1 eV) con Au como electrodo (T, O y O p denotan tiofeno, tiofeno-1,1-dióxido y tienopirrolodiona oxidada, respectivamente) [45]. Los resultados muestran que el nivel de Fermi de Au está en el medio de LUMO y HOMO. Por lo tanto, podemos inferir que el nivel de Fermi de Au puede estar alrededor del nivel de energía promedio de LUMO y HOMO, que son - 4.25 y - 4.35 eV establecidos a partir de TOTOT y TTO P TT, respectivamente. El nivel de Fermi de Au - 4,25 y - 4,35 eV es similar al medido por UPS con - 4,2 eV [43]. De acuerdo con lo anterior, usaremos el - 4.2 eV como el nivel de Fermi del electrodo de Au con la adsorción de la molécula.
Suponiendo que el nivel de Fermi de - 4.2 eV para Au con SAM, C6DT y C6DA son el transporte de electrones dominado por HOMO, mientras que el transporte de electrones dominado por LUMO se propone para el C6DC y C6DI. Por lo tanto, la altura de la barrera Φ puede establecerse como 1,98 eV (C6DT), 1,82 eV (C6DA), 1,62 eV (C6DC) y 1,59 eV (C6DI) (Tabla 1). La tendencia de la altura de la barrera entre la molécula y Au es Φ C6DT (tiol)> Φ C6DA (amina)> Φ C6DC (ácido carboxílico)> Φ C6DI (yodo), que es consistente con la tendencia de la desintegración por efecto túnel ( β ). Por lo tanto, la inusual baja desintegración de túnel puede contribuir a la pequeña altura de la barrera entre las moléculas de alcanos terminadas en yodo y el Au.
Conclusiones
En conclusión, hemos medido la conductancia de moléculas basadas en alcanos con el contacto del grupo yodo con los electrodos de Au por STM-BJ a temperatura ambiente. Una decadencia tuneladora β N de 0,5 por –CH 2 se encontró para aquellas moléculas con electrodos de Au, que es mucho más bajo que el de las moléculas a base de alcanos con otros grupos de anclaje. Esto puede deberse a la pequeña altura de la barrera entre la molécula de alcano terminada en yodo y el Au. El trabajo actual muestra el importante papel del grupo de anclaje en las características eléctricas de las uniones moleculares individuales, que pueden sintonizar la desintegración de túnel de la unión molecular y guiar la fabricación del cable molecular.
Abreviaturas
- HOMO:
-
Orbital molecular más alto ocupado
- LUMO:
-
Orbital molecular desocupado más bajo
- SAM:
-
Monocapas autoensambladas
- STM-BJ:
-
Microscopía de túnel de barrido rompe uniones
- UPS:
-
Espectroscopia de fotoelectrones ultravioleta
Nanomateriales
- El Consejo Europeo de Investigación financia la investigación de dispositivos de una sola molécula mediante la manipulación de átomos
- Efecto superficial sobre el transporte de petróleo en nanocanales:un estudio de dinámica molecular
- Propiedades de fotoluminiscencia de modificaciones polimórficas de poli (3-hexiltiofeno) de bajo peso molecular
- Modelado y simulación de dinámica molecular del corte con diamante de cerio
- Dependencia de la temperatura de la banda prohibida en MoSe2 crecido por epitaxia de haz molecular
- Electrodo de puerta plateado impreso con inyección de tinta y curado por UV con baja resistividad eléctrica
- Láseres de cascada cuántica DFB de bajo consumo de energía
- Estudio del efecto de la dirección de impacto en el proceso de corte nanométrico abrasivo con dinámica molecular
- Estudio de los comportamientos de fricción a nanoescala del grafeno en sustratos de oro utilizando dinámica molecular
- Síntesis y estudio in vitro de una sonda de modo dual que apunta a la integridad αvβ3
- Causas del factor de potencia bajo