


Nanodiamantes emisores de verde de alta calidad fabricados mediante sinterización HPHT de diamantes de onda de choque policristalinos
Resumen
Demostramos una técnica de sinterización de alta presión y alta temperatura para formar centros de nitrógeno-vacío-nitrógeno en nanodiamantes. Los precursores de nanopartículas de diamante policristalino, con un tamaño medio de 25 nm, son producidos por la onda de choque de una explosión. Estas nanopartículas se sinterizan en presencia de etanol, a una presión de 7 GPa y una temperatura de 1300 ° C, para producir cristalitos de diamante sustancialmente más grandes (3-4 veces). Las propiedades espectrales registradas demuestran la calidad cristalina mejorada. También se observa que cambian los tipos de defectos presentes; desaparecen los rasgos espectrales característicos de los centros de vacantes de nitrógeno y de silicio presentes para el material precursor. Aparecen dos nuevos rasgos característicos:(1) nitrógeno sustitutivo paramagnético (centros P1 con espín ½) con una estructura hiperfina triplete característica de resonancia paramagnética de electrones debido a la I =1 momento magnético del espín nuclear del nitrógeno y (2) la firma de fotoluminiscencia espectral verde de los centros de nitrógeno-vacío-nitrógeno. Este método de producción es una fuerte alternativa a la irradiación convencional con haz de partículas de alta energía. Se puede utilizar para producir fácilmente nanodiamantes fluorescentes puramente verdes con propiedades ventajosas para aplicaciones de bioetiquetado óptico.
Introducción
Las impurezas y las vacantes de nitrógeno son los defectos predominantes en la mayoría de los diamantes naturales y sintéticos. Los defectos individuales pueden formar colectivamente complejos de defectos con hasta 6 subunidades [1, 2]. De estos complejos de defectos, nitrógeno-vacante (NV - ) y, en menor medida, los centros de nitrógeno-vacío-nitrógeno (NVN) han atraído un interés significativo, debido a su fotoluminiscencia roja y verde que no parpadean, respectivamente [3, 4]. NV - y NVN se puede generar de forma controlable en nanodiamantes. Los nanodiamantes son ampliamente reconocidos como nanopartículas no tóxicas y, por lo tanto, pueden usarse como etiquetas trazables a largo plazo en aplicaciones biomédicas [5]. Nanocristales con NV - Los centros de color también se utilizan para la detección cuántica [3].
La síntesis de diamantes a alta presión y alta temperatura (HPHT) con catalizadores solventes de metales de transición convencionales es una técnica industrial estándar. Se explota en muchos laboratorios para cultivar cristales de diamante con parámetros de celosía avanzados. Sin embargo, en los últimos años se han comenzado a utilizar ampliamente varios catalizadores metálicos no convencionales [6]. Este método permite el uso de aditivos orgánicos que contienen nitrógeno y absorbentes de metales para dopar de manera controlada el diamante en un rango que va desde un alto contenido de nitrógeno (hasta ~ 1000 ppm), hasta un nivel mucho más bajo (~ 50 ppm) [7, 8]. . El HPHT también se usa para templar cristales de diamante y mejorar su calidad cristalina, para decolorarlos y para sinterizar nanocristales en policristales más grandes.
La agrupación de defectos de nitrógeno individuales en complejos más grandes en el diamante bajo la influencia de la temperatura (y la presión) se ha estudiado extensamente. Estos complejos pueden servir como marcadores específicos, registrando el historial de temperatura de los procesos de cristalización completados [1]. NVN y NV - Los centros se pueden crear irradiando diamantes prístinos que contienen nitrógeno con electrones o protones de alta energía (2-14 meV), o iones pesados de ~ 40 keV. La irradiación crea vacíos en la red de diamantes. El recocido posterior de las muestras a temperaturas en el rango de 500-2000 ° C provoca la agrupación de los defectos [3, 9, 10]. Actualmente, la producción en masa de NV - o los nanodiamantes que contienen NVN se obtienen típicamente mediante irradiación con electrones de alta energía. Sin embargo, este proceso es caro y requiere mucho tiempo y requiere una preparación específica de la muestra. Por lo tanto, vale la pena desarrollar métodos alternativos para la creación de centros de color en nanodiamantes, que no requieren irradiación de partículas.
Recientemente se introdujo un nuevo método para fabricar cristales de diamante de tamaño submicrónico, que se basa en la sinterización de nanodiamantes de detonación (DND) de 5 nm en presencia de aditivos líquidos C – H – O [11, 12]. En condiciones de sinterización HPHT, los compuestos orgánicos C – H – O añadidos se comportan como un fluido supercrítico que induce una rápida recristalización del diamante. La rápida recristalización va acompañada de una mayor densidad de vacantes [13]. Este método, sin embargo, suprime los centros de color prístinos presentes en los DND precursores de 5 nm [14]. Por lo tanto, es deseable un enfoque alternativo que pueda preservar estos defectos o incluso crear otros tipos durante el proceso de sinterización HPHT.
En este trabajo, utilizamos nanodiamantes policristalinos precursores producidos por un método asistido por explosión utilizando la llamada tecnología de síntesis de ondas de choque de DuPont [15,16,17]. Sinterizamos estos nanodiamantes en presencia de aditivos C – H – O de la misma manera que en un procedimiento patentado existente [18] (Fig. 1), obteniendo diamantes submicrónicos gruesos que contienen solo centros de color NVN, que no estaban presentes en el precursor. nanopartículas. También realizamos un seguimiento de la evolución de todas las NV - , P1 y SiV se centra en las partículas antes y después de la sinterización, utilizando resonancia paramagnética electrónica (EPR) y fluorescencia para demostrar el efecto de la sinterización en la gama completa de defectos presentes.
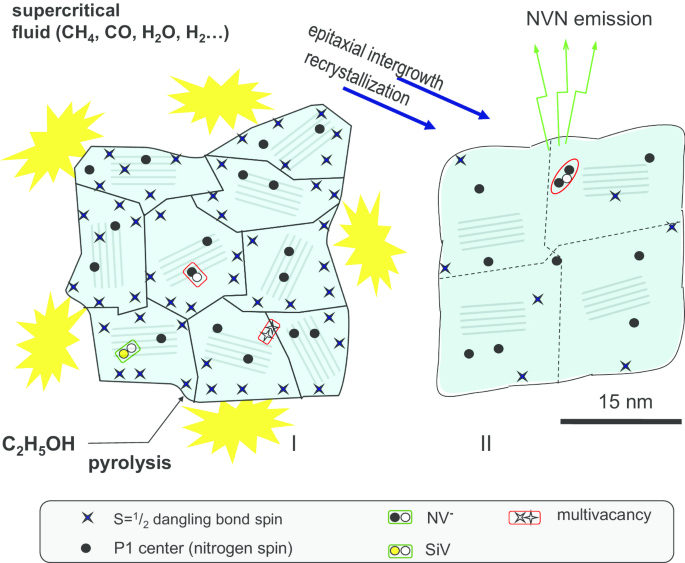
Ilustración esquemática de la sinterización de partículas de diamante policristalino en condiciones de alta presión y alta temperatura para producir nanocristales de diamante submicrónicos con tamaños de aproximadamente 30 nm. Se observan diversos defectos y centros de color tanto en policristales (I) como en nanocristales tras la sinterización HPHT (II)
Métodos
Fabricación de muestra
Los cristales de diamante submicrónicos se prepararon sinterizando nanodiamantes policristalinos precursores disponibles comercialmente con un tamaño medio de 25 nm (producto DP 0-0,05, Microdiamant, Suiza). Las partículas precursoras se obtienen mediante una técnica asistida por explosivos en la que se produce una conversión de grafito a diamante rápida, inducida por ondas de choque (dentro de cien microsegundos en T ≈ 950 ° C, P ≈ 50 GPa). Los productos resultantes de la explosión se tratan posteriormente químicamente para depurar la fase de nanopartículas de diamante, eliminando los metales residuales y el carbono amorfo. El polvo estaba compuesto por grandes policristales con tamaños> 10 mμ. Estos policristales se pueden fraccionar fácilmente, moliendo y desagregando a lo largo de los límites de los granos, siguiendo un método descrito anteriormente [19]. Se llevó a cabo el fraccionamiento por tamaño para extraer la fracción de tamaño pequeño que forma los policristales de DP 0-0,05. La distribución de tamaño resultante (Fig. 2) para la fracción más pequeña tiene su máximo a 27 nm y una mitad máxima de ancho completo (FWHM) ≈ 25 nm (con el 95% de las partículas con tamaños en el rango de 3 a 50 nm). Las partículas precursoras seleccionadas se transfieren luego al cilindro de grafito interior de una cámara de alta presión de tipo toroidal para la sinterización. Los tamaños típicos de la parte interior de la cámara de alta presión (es decir, el cilindro de grafito hueco mencionado anteriormente con dos tapas de grafito) fueron:diámetro interior 4,0 mm y altura 5,5 mm. Antes de la sinterización, se añadió etanol gota a gota al polvo de nanodiamantes secos hasta llenar completamente el espacio entre partículas (alrededor del 30-50% en peso) [18]. Luego, la sinterización tuvo lugar en condiciones de alta presión (7 GPa) y alta temperatura (1300 ± 50 ° C) durante 10 s. Durante la ejecución de una prensa, se podrían tratar aproximadamente ~ 120 mg de polvo de diamante precursor. El esquema de la cámara de alta presión se puede encontrar en la Ref. [11].
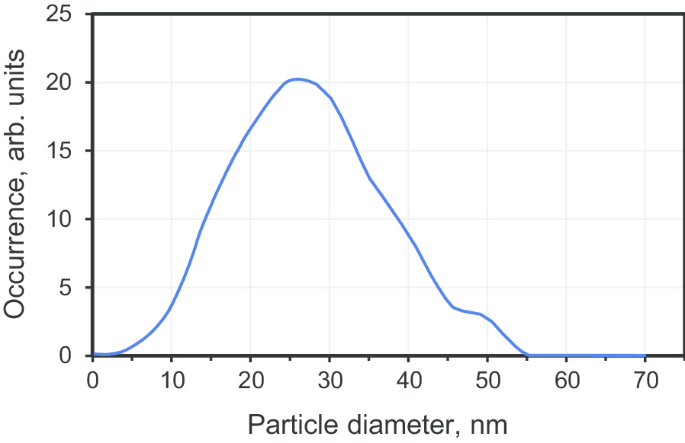
La distribución de tamaño de la fracción de diamante policristalino DP 0-0,05 con un tamaño medio ~ 27 nm medido por un aparato de sedimentación centrífuga diferencial (centrífuga de disco CPS, CPS Instruments Inc., EE. UU.)
El efecto de este proceso de sinterización se ilustra en la Fig. 1. Cuando se sinteriza en condiciones HPHT, el etanol se encuentra en un estado supercrítico. Por lo tanto, puede penetrar fácilmente los límites de los granos policristalinos, promoviendo la recristalización del diamante y el crecimiento de una nueva fase de diamante. Después de la sinterización, se extrajeron ~ 90-100 mg de un polvo de diamante blanco de calidad aceptable (sin contaminación del recipiente de grafito) de la cámara de alta presión (etiquetada como D19 en este documento). El cambio de color del polvo, del gris / negro del precursor, al blanco después de la sinterización indica un cambio en la superficie de los policristales y su correspondiente crecimiento [11].
Caracterización estructural de los materiales antes y después de la sinterización
Caracterización estructural del precursor de nanodiamantes policristalinos
Primero caracterizamos la estructura de DP 0-0.05 antes de sinterizar. Las mediciones de difracción de rayos X (XRD) se tomaron con un difractómetro de rayos X Rigaku Smart Lab III, utilizando radiación CuKα ( λ =1,54178 Å). Se utilizaron una tensión de 40 kV, una corriente de 30 mA y una velocidad de exploración de 0,1 grados / min. El patrón XRD de DP 0-0.05 se muestra en la Fig. 3 (negro) para 2 θ en el rango de 6 ° a 155 °. Observamos seis picos, cinco de los cuales (111, 220, 311, 400 y 331) son característicos de la fase diamante del carbono. El pico más fuerte observado es el diamante cúbico (D c ) 111 en 2 θ ≈ 43,7 °. Este pico no es simétrico, presentando un hombro izquierdo. Ajustar el patrón con dos Lorentzianos produce un primer pico centrado en 2 θ =43,76 ° y un segundo a 2 θ =41,64 °. El primer pico corresponde a la reflexión de (111) planos de diamante, mientras que el segundo (etiquetado D H 011) se atribuye tentativamente a:(a) reflejos de planos de una fase de diamante politipo 6H hexagonal [20] oa (b) múltiples fallas de apilamiento, gemelos y límites de grano relacionados entre los cristalitos bajo tensión alta. Comparando las integrales de D H 011 y D c 111 picos, estimamos que la "fase" no identificada (ya sea hexagonal o compuesta de defectos estructurales) en el precursor DP 0-0.05 para representar ≈ 20% en peso. El pico etiquetado 002 Graphite (a 2 θ ≈ 26 °) es atribuible a una fase de nanografito. Si bien esta fase de impureza se puede eliminar fácilmente, no grabamos intencionalmente con ácido en este estudio. Comparando las áreas de los picos 002 y 111, se evaluó que esta fase amorfa contribuía con ~ 4% en peso. Luego evaluamos la longitud de la región de dispersión coherente ( L CSR ) utilizando el método Scherer. Consideramos el FWHM β de los cinco picos de diamantes en función de sec θ . Β se determina después de la desconvolución de la función de respuesta del difractómetro. Nota al pie 1 La L CSR para la muestra de DP 0-0,05 fue de 6,7 nm.
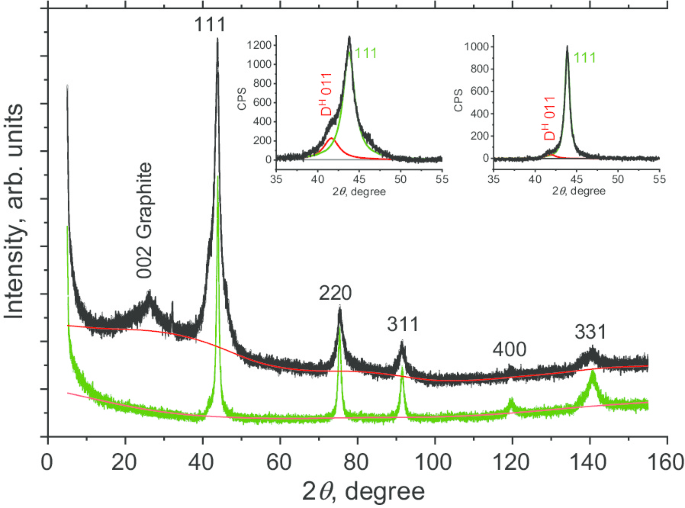
Perfiles de XRD en polvo de la fracción DP 0-0,05 de partículas de diamante policristalino (curva negra) y nanodiamantes D19 obtenidos por sinterización (curva verde). Recuadros:ajuste de la reflexión más intensa de los planos (111) con dos curvas Lorentzianas (rojo y verde) centradas en 41,5 ° (D H 011) y 43,8 o (D c 111) para muestras antes (recuadro izquierdo) y después de la sinterización (recuadro derecho)
Para obtener más información sobre la complicada estructura policristalina del diamante precursor DP 0-0.05, utilizamos un microscopio electrónico de transmisión de alta resolución (HRTEM, JEOL JEM-2100F equipado con correctores CEOS Cs, Universidad de Shinshu). El HRTEM se hizo funcionar a 80 kV para minimizar el daño por irradiación. Las imágenes HRTEM (Fig. 4) muestran que los policristales están compuestos de cristalitos de nanodiamantes cúbicos estrechamente unidos con la característica 1,93 Å d -flecos espaciadores. El policristal fotografiado en la Fig. 4a está compuesto de cristalitos con tamaños de 5 a 12 nm, que tienen diferentes orientaciones marcadas por diferentes colores. Además, HRTEM demuestra que el tamaño de los policristales individuales que componen el polvo de DP 0–0,05 está ampliamente distribuido, en el rango de 5–50 nm. Las imágenes HRTEM no revelaron ningún rastro de una nanofase de diamante hexagonal (como la lonsdaleita). Sin embargo, observamos muchos límites de hermanamiento cortos (Fig. 4c, d), algunos de ellos exhibiendo planos de celosía plisada con formas de acordeón como en la Fig. 4d. Estas observaciones indican que el hombro del pico de 111 XRD (Fig. 3) discutido anteriormente probablemente se deba a fallas de apilamiento asociadas con múltiples límites de grano relacionados con el hermanamiento más que a la presencia de una fase de diamante hexagonal [16].
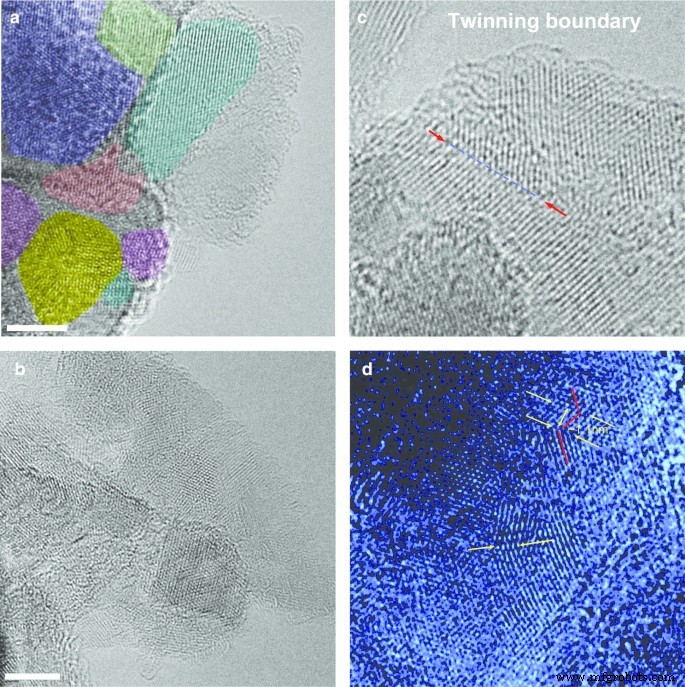
Imágenes TEM de alta resolución de policristales de diamante cúbicos seleccionados extraídos del polvo DP 0-0.05 ( a , b ) e imágenes típicas de los límites de hermanamiento simple, de varios nanómetros de longitud, que se encuentran ocasionalmente en la muestra ( c , d ). Paneles ( a , b ):barra de escala — 4 nm. Los diferentes cristalitos se destacan en diferentes colores en ( a ). Flechas en paneles ( c ) y ( d ) marcar los límites de hermanamiento seleccionados claramente distinguibles
La composición elemental de DP 0-0.05 se analizó mediante el método Pregl-Dumas utilizando un sistema de análisis elemental orgánico (JM10, J-Science Lab Co., Ltd, Kyoto, Japón) en el que la muestra se quemó a 1007 ° C bajo un flujo de oxígeno (30 ml / min). La fracción de oxígeno se determinó mediante una balanza. Los resultados del análisis, en% en peso, son:C — 90,45, N — 2,47, H — 0,76, O — 6,32. La concentración relativamente grande de nitrógeno significa que casi todo el nitrógeno está presente dentro de los policristales en forma agregada, probablemente en forma de centros A (dímeros NN). Un análisis adicional por fluorescencia de rayos X mostró la presencia de otros oligoelementos en DP 0-0.05:Fe (~ 300 ppm), Cu (35 ppm), Si (120 ppm), Cr (~ 150 ppm), Ca (~ 45 ppm), Mn (45 ppm), P (30 ppm), Al (18 ppm), Ti (13 ppm), Mg (6 ppm), Ni (2 ppm), Zn (2 ppm), Co (1 ppm) .
La muestra de DP 0-0.05 utilizada para estudios posteriores de resonancia magnética se trató adicionalmente en ácido clorhídrico hirviendo para reducir la presencia de metales ferromagnéticos (principalmente hierro y cromo) hasta el nivel de ~ 10 ppm.
Caracterización estructural del diamante sinterizado
También caracterizamos de manera similar la muestra D19 obtenida sinterizando DP 0-0.05. El patrón D19 XRD se obtuvo por el mismo método que para el polvo DP 0-0.05. Se encontró que mostraba solo los cinco picos de Lorentz característicos de una fase de diamante cúbico (Fig. 3, línea verde) correspondientes a los planos 111, 220, 311, 400 y 331. No hay evidencia de la fase de grafito presente en el material precursor. La disimetría de 111 picos también está fuertemente reducida (Fig. 3, recuadro derecho). Esto sugiere una reducción sustancial correspondiente en las fallas de apilamiento. Además, la L CSR Se encontró que la longitud de coherencia era de 10,8 nm, lo que indica el agrandamiento de los nanocristalitos de diamante durante la sinterización. Teniendo en cuenta estas observaciones, planteamos la hipótesis de que las fases de diamante grafítico y no cúbico se convierten en la fase de diamante cúbico durante el proceso de sinterización. Esto probablemente sea el resultado de la disolución de estas fases, seguida del crecimiento de una fase cúbica de diamante dentro de los espacios intersticiales entre los cristalitos de los policristales. Este proceso se ha observado anteriormente para los nanodiamantes de detonación [14].
Las imágenes de microscopía óptica y electrónica de barrido de la muestra D19 se presentan en la Fig. 5a, b, respectivamente. Aquí, vemos las partículas de forma arbitraria con tamaños de ~ 0.3 a ~ 5 micrones. En realidad, son agregados densos submicrométricos y muy sueltos de tamaño micrométrico de cristalitos de diamante mucho más pequeños unidos por enlaces covalentes o enlaces débiles de Van der Waals. Dichos enlaces suelen resultar de las interacciones de los grupos funcionales superficiales de las partículas de diamante vecinas. Las partículas de diamante individuales que constituyen estos agregados sueltos pueden verse mediante microscopía electrónica de transmisión. En la figura 5c se muestra una imagen TEM de un cristalito de diamante D19 con un tamaño de aproximadamente ~ 100 nm. Esta imagen se tomó con un microscopio electrónico de transmisión JEOL JEM-2100F (Universidad de Hosei) a un voltaje de aceleración de 200 kV. La muestra se fijó en una rejilla de cobre sin sustrato de carbono.

Óptico ( a ), microscopía electrónica de barrido ( b ) y microscopía electrónica de transmisión ( c ) imágenes de las partículas de diamante D19 sintetizadas. La imagen óptica se tomó con un objetivo microscópico de 100x. Los parámetros correspondientes utilizados para tomar la imagen electrónica SEM se indican en la parte inferior del panel ( b )
Métodos para analizar el contenido en defectos de nanodiamantes pre y post-sinterizados
Ambas muestras de diamantes fueron analizadas mediante EPR, Raman y espectroscopias de fluorescencia. Los espectros EPR se registraron a temperatura ambiente, a una frecuencia de microondas de 9,444 GHz, utilizando un espectrómetro EPR (JES-FA 300, JEOL, Japón). Se introdujo una masa de 20 mg de polvo en un tubo EPR de cuarzo de 4 mm de diámetro. La altura de la columna de polvo en el tubo no superó los 10 mm. El extremo abierto del tubo se selló contra la humedad.
Espectros EPR con g -factores en el rango g =4,00–4,30 se registraron con una potencia de microondas de P MW =10 mW, amplitud de modulación del campo magnético A m =1 mT y frecuencia ν =100 kHz, ganancia del amplificador G ≈ 10 3 y N =16 ciclos de acumulación de señal. Estos parámetros se eligieron para obtener la relación señal-ruido óptima. La constante de tiempo fue de 0,03 s, y el tiempo total de registro del barrido del campo magnético durante el intervalo de 130 a 200 mT fue de 120 s. Espectros EPR con g -factores g ≈ 2 se registraron en el intervalo de 327 a 347 mT, con una potencia de microondas de P MW =0,03 mW, amplitud de modulación del campo magnético A m =0.035 mT, ganancia del amplificador G ≈ 10 2 y N =4 ciclos de acumulación de señal. Tenga en cuenta que, como regla general, para las principales señales EPR amplias ( g ≈ 2) con un ancho de línea> 0,5 mT, la intensidad de la señal EPR de pico a pico ( I pp ) sigue una dependencia de potencia de MW I pp ~ ( P MW ) 1/2 hasta P MW ≈ 100 mW. Por otro lado, para señales EPR estrechas (anchos de línea <0,15 mT a baja potencia), I pp satura en P MW > 0.05 mW y tiene una fuerte distorsión de forma a valores más altos (> 4 mW). Estas tendencias de saturación se observaron tanto para el precursor policristalino como para los diamantes sinterizados posteriormente.
Adquirimos fotoluminiscencia (PL) y espectros Raman, con un espectrómetro micro-Raman (“inVia”, Renishaw, Reino Unido), junto con un microscopio óptico (Leica, Alemania) utilizando un objetivo de 50 × (NA =0,78) y un CCD detector enfriado a -70 ° C, en una geometría de retrodispersión. Los espectros se registraron con una resolución espectral de ~ 2 cm –1 . Usamos las dos líneas láser de un láser de iones de argón a 488 nm y longitudes de onda de 457 nm, con intensidades inferiores a 20 W cm −2 en el punto focal de la muestra. Registramos imágenes espectrales en el modo StreamLine ™ Plus (Renishaw, Reino Unido) que utiliza una intensidad de láser de excitación más baja en el plano de la muestra debido a su enfoque en una franja de 2 × 30 mμ, en comparación con el enfoque estándar en un círculo de tamaño micrón. Esta estrategia limitó el daño de la muestra inducido por láser y el grabado local en fase gaseosa a través del sobrecalentamiento y la oxidación en la atmósfera ambiental. Previamente se han descrito más detalles de esta técnica y del prensado de polvo de diamante en cilindros de 2 mm de diámetro [19].
Se obtuvieron imágenes fluorescentes de partículas D19 aisladas con microscopía confocal de epifluorescencia de campo amplio con un objetivo de 100x. Las partículas se depositaron en un cubreobjetos de vidrio, a partir de la fracción sobrenadante de una suspensión diluida a base de agua de polvo D19, mediante recubrimiento por centrifugación. El cubreobjetos se trató preliminarmente en plasma de oxígeno para evitar la fluorescencia parásita de los orgánicos residuales y promover una mejor unión de las partículas D19 al cubreobjetos. La fluorescencia se excitó con un láser de 488 nm (potencia de excitación ≈ 40 mW) y se recogió con un filtro de interferencia 525/40. Se utilizó un detector CCD de matriz 2D especialmente enfriado (temperatura de la cámara =- 79,9 ° C) para registrar las imágenes. Las imágenes (tamaño ~ 80 × 80 μm, 80,00 nm / píxel) se registraron con un tiempo de exposición de 60 ms. Se utilizó una paleta en escala de grises para la presentación de imágenes. La imagen se analizó con el software Fiji.
Resultados
EPR de policristales y cristalitos de diamante antes y después de la sinterización
Los espectros de EPR del polvo de diamante policristalino DP 0–0,05, tanto en el rango de campo medio magnético como en el rango de campo alto, se muestran en la Fig. 6. Los espectros de EPR registrados en estos distintos rangos se midieron en microondas alto y bajo poderes P MW =10 mW y P MW =0,03 mW, respectivamente. Aquí, la región de campo alto se seleccionó normalmente en la vecindad de la línea de absorción relacionada con el flip-flop principal inducido por microondas Δ M s =1 transiciones de giros S =½, mientras que el rango de medio campo extendido se seleccionó especialmente para buscar las señales de Δ M s =2 transiciones de posible triplete S =1 centros. El espectro EPR de medio campo tiene una intensidad muy baja y demuestra la presencia de triplete NV - centros (señal con g =4.27 en H res =158 mT [10, 16]) y multivacancias de triplete (señal con g =4,00 a las H res =168 mT [16]) en el precursor de diamante policristalino. Ambos tipos de defectos están presentes en concentraciones muy pequeñas (<1 ppm); por lo tanto, fue necesario utilizar una alta potencia de microondas para detectar centros tan raros. Además, en el dominio de campo alto también observamos la firma característica de los centros de la mitad de espín ( g =2.0027 en H res =337 mT [10, 16, 21]), pero con un ancho (Δ H pp =0,47 mT), forma única derivada de Lorentzian sin ninguna estructura fina [16]. Dada la suposición de que estos espines provienen de centros independientes, esta señal amplia e intensa puede atribuirse tentativamente a espines de enlaces colgantes C – C y al intercambio de espines de nitrógeno paramagnético acoplados con estructura hiperfina no resuelta (HFS). Nota al pie 2 Estimamos la concentración de todas las especies paramagnéticas con espín S =1/2 para ser ~ 4 × 10 19 spin / g (800 ppm), que es ~ 1,5 veces más pequeño que el valor previamente informado para los nanodiamantes de detonación de 5 nm [22].
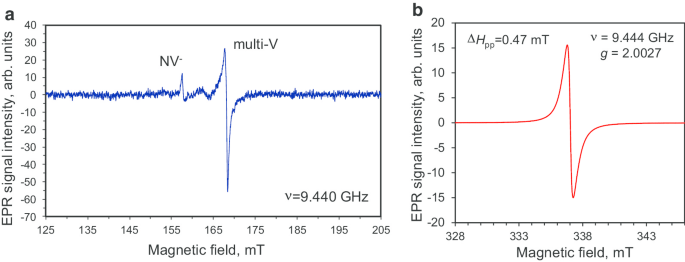
Espectros EPR de la fracción DP 0-0,05 de partículas de diamante en el rango de medio campo magnético ( a ) y alrededor del campo magnético resonante del singlete con una señal fuerte en g -factor g ≈ 2.0027 ( b ). Potencia de microondas P MW :10 mW ( a ) y 0,03 mW ( b ). Frecuencia de microondas ν =9,44 GHz. Ambos espectros se registran en el régimen lejos de la saturación
La Figura 7a muestra los espectros EPR de la muestra D19 para muy baja ( P MW =1 μW) y potencias de microondas muy altas (200 mW), es decir en los regímenes muy por debajo de la saturación o en la saturación (como lo demuestra el ensanchamiento), respectivamente. El espectro de P MW =1 Wμ tiene una línea central con un ancho de línea más estrecho de 0.14 mT en comparación con el de las partículas precursoras. La Figura 7b (curva 1) muestra la señal EPR de baja potencia de microondas doblemente integrada. Esta señal se puede descomponer en:una señal de forma Lorentziana amplia (FWHM ≈ 1,7 mT) (Fig. 7b, curva 2) asociada con la S más abundante =1/2 especie (de alta densidad local y organizada en grupos de espines) y una línea central muy estrecha con dos líneas satélites simétricas separadas por ~ 6 mT (Fig. 7b, curva 3). Las relaciones de las intensidades integradas para las dos líneas de satélite en relación con la línea central son 0,90 y 1,09, respectivamente. Por lo tanto, tienen intensidades integradas iguales con una precisión de medición experimental de ± 10%. Las líneas de satélite están relacionadas con la estructura hiperfina de la señal EPR del nitrógeno sustitutivo paramagnético neutro ( 14 N, S =1/2, yo =1), conocidos como centros P1 [23, 24]. No se observaron centros P1 en los nanodiamantes policristalinos precursores, pero se detectan claramente en el diamante después de la sinterización. Trabajos anteriores han demostrado que esta estructura característica triplete HFS relacionada con los centros P1 aparece sólo en cristales de diamante gruesos con un tamaño superior a 50-80 nm [25]. Además, se ha demostrado que la sinterización de nanodiamantes de detonación en presencia de etanol promueve el aumento de cristalito basado en la recristalización del diamante y el crecimiento de la fase de diamante nuevo [26]. Por lo tanto, los datos actuales de EPR están de acuerdo con una ampliación similar que ocurre aquí desde cristalitos individuales de partículas policristalinas con un tamaño de ~ 7-10 nm a nuevos cristalitos de tamaño individual ~ 40-50 nm. Nota al pie 3
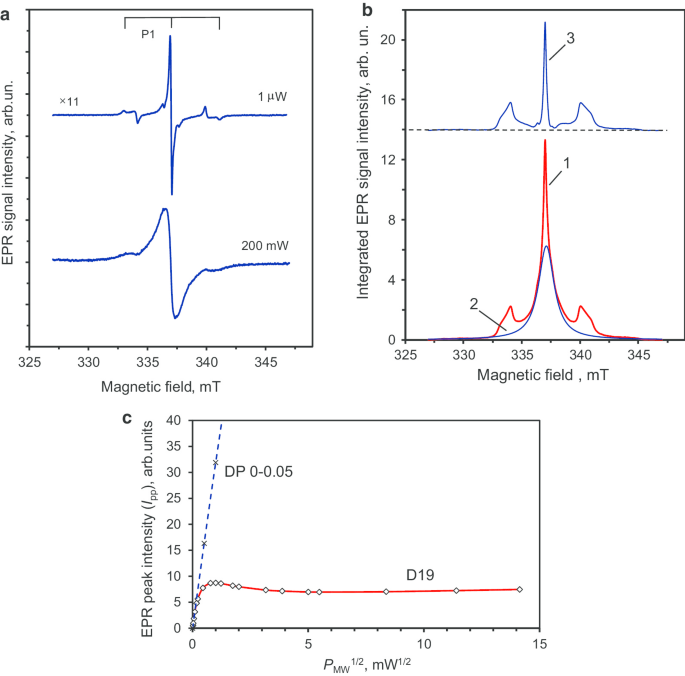
La principal señal EPR de primera derivada de los cristales de diamante D19 a potencias de microondas altas y bajas ( a ), descomposición de la señal EPR integrada en componentes relacionados con dos grupos de espines ( b ) y la tendencia de saturación correspondiente de la intensidad máxima de esta señal EPR frente a la raíz cuadrada de la potencia de microondas en el rango de hasta 200 mW ( c ). La estructura triplete HFS de la señal EPR del centro P1 (nitrógeno de sustitución) se distingue claramente en ( a ). En ( b ):la curva 3, que tiene estructura de triplete, corresponde al espectro EPR de solo centros P1. En ( c ):la línea recta discontinua es la I pp vs ( P MW ) 1/2 dependencia para policristales DP 0-0,05 dada como referencia. Cuatro puntos experimentales en P MW =0.5, 1, 2, 4 mW se utilizaron para su trazado (dos de ellos no se presentan aquí). Frecuencia de microondas ν =9,44 GHz
Esta hipótesis se confirma no solo por la observación de la señal EPR estrecha Footnote 4 ( g =2.0024) de los centros P1 con características HFS [26], sino también por la detección de I pp saturación por encima de P MW =0,7 mW para la muestra D19. Para el nanodiamante policristalino precursor DP 0–0,05, la intensidad máxima de la señal EPR principal no se satura incluso a un alto nivel de potencia de microondas ( P MW =20 mW). La dependencia del poder I pp ~ ( P MW ) 1/2 se mantiene bien en todo el rango de potencia de microondas ( P MW =0-20 mW). El yo pp ~ ( P MW ) 1/2 La dependencia para la muestra de DP 0-0,05 se muestra en la figura 7c mediante una línea discontinua, hasta ~ 16 mW de potencia de microondas. Tal dependencia lineal sin saturación resulta de la alta concentración de centros paramagnéticos dentro de los nanodiamantes policristalinos y los tiempos de relajación de espín-rejilla y espín-espín muy cortos. El comportamiento de saturación de la muestra D19 g =2.0024 La señal EPR también se muestra en la Fig. 7c en el rango de 0-200 mW. Nota al pie 5 Observamos un I pp ~ ( P MW ) 1/2 dependencia solo para potencia de microondas por debajo de ~ 15 Wμ. En este caso, yo pp demuestra saturación en el rango por debajo de 1 mW y alcanza un máximo en ~ 1 mW. El yo pp luego disminuye sustancialmente en el rango de 1 a 25 mW antes de volver a aumentar lentamente hasta la potencia máxima utilizada (200 mW). La presencia de tal tendencia de saturación en I pp ( P MW ), y también la caída sobre P MW =1 mW, es característico de los centros P1 con tiempos de relajación espín-espín y espín-retículo relativamente grandes, ubicados lejos de otros defectos y de los bordes de las partículas [27]. Estas condiciones se cumplen para concentraciones centrales P1 inferiores a 200 ppm en nanodiamantes tipo Ib HPHT con un tamaño superior a 60-80 nm. Sin embargo, el tamaño medio real de los cristalitos de diamante D19 individuales (partículas elementales), como se ve solo desde el punto de vista de EPR, es una cuestión abierta. Puede resolverse a grandes rasgos comparando el espectro de EPR real de D19 con la serie de espectros de EPR de polvo de diamantes Ib HPHT molidos con un tamaño medio que varía de 18 a 390 nm [28]. Siguiendo la Ref. [28], donde se publicaron estos espectros EPR, las firmas P1 HFS relacionadas con el nitrógeno de sustitución están completamente ausentes en los diamantes en polvo con un tamaño medio ≤ 30 nm, pero todavía están presentes en las muestras de tamaño intermedio (85-130 nm). Esta comparación indica que el tamaño medio de los cristalitos de diamante D19 sintetizados está en la región de 10.030 ± nm. This estimation coincides well with the representative size observed in the TEM image shown in Fig. 5c. It is notable that the EPR spectrum of D19 recorded at the high power of P MW = 200 mW (Fig. 7a) shows a lack of definition of the HFS structure of the P1 signal. The broad central line suggests the presence, at the nanoscale, of dense clusters of paramagnetic spin-half that strongly couple to each other.
Altogether, the decrease in the linewidth of the main EPR signal (g = 2.0024), the appearance of well-defined HFS characteristics in the P1 centre spectrum after sintering, and the I pp saturation for P MW > 0.7 mW (Fig. 7c) are suggestive of an increase in size by up to one order of magnitude (crystal size > 50 nm). It also indicates a better crystallinity of the nanodiamond in the D19 sample. From van Wyk measurements [29], a smaller amount of paramagnetic defects (< 200 ppm) are expected, based on the narrow linewidth (0.14 mT) of the g = 2.0024 main paramagnetic signal in the D19 sample.
Fluorescence and Raman Scattering of Diamond Crystals
The photoluminescence (PL) spectrum of the DP 0–0.05 precursor together with the PL spectra of some much coarser fractions of polycrystalline diamond particles (DP 0–0.2 and DP 0–0.35) produced by Microdiamant TM is shown in Fig. 8. The spectrum of DP 0–0.05 under the 488 nm excitation wavelength has two features of note:the prominent narrow PL line at 738 nm, associated with the zero-phonon line of negatively charged SiV − centres, and a broad spectrum background with PL bands centred at 525, 600, 660 and 740 nm, associated with various light-emitting centres in diamond, including NV centres. For polycrystals with mean size 25 nm (DP 0–0.05), the intensities of these bands are smaller than that for polycrystals with mean sizes of 100 and 175 nm (DP 0–0.2 and DP 0–0.35, respectively). A more detailed analysis of the PL spectra of polycrystalline DP 0–0.05 particles has been previously undertaken [19].

PL spectra of various submicron fractions of Microdiamant™ polycrystalline diamond particles:blue—DP 0–0.05 (mean size 25 nm), green—DP 0–0.2 (mean size 100 nm), red—DP 0–0.35 (mean size 175 nm). Excitation wavelength λ = 488 nm. The prominent peak at 738 nm marked by the vertical dashed line is the zero-phonon line of negatively charged SiV − centres, which can be observed in all polycrystalline diamond fractions. For better comparison, the spectra are specially normalized for PL intensity at λ = 590 nm. Normalising coefficients are indicated in the figure
Figure 9a shows the PL spectrum of sintered diamond sample D19 (blue curve) at room temperature (RT) together with the PL spectrum of the DP 0–0.05 precursor (red curve). The D19 spectrum displays a green fluorescence characteristic, with a sharp maximum at 525 nm, and a subsequent decrease at larger wavelengths. Note that the single-phonon, sharp Raman line of diamond, which is expected at 522 nm, was too weak to be detected on the ascending slope of the PL signal under 488 nm excitation wavelength. Footnote 6 As previously reported [9, 30], such spectra—with a continuous higher wavelength band of “triangular” shape—are characteristic of optical emission from NVN centres (also known as H3 centres) in submicron (< 140 nm) diamonds at RT. In the PL spectrum of sample D19, at least four broad bands (“bumps”) centred at 538, 569, 601 and 710 nm can be additionally distinguished. We do not believe that they are related to phonon sidebands of the NVN (H3) centres. The origin of the 525 nm sharp peak and “bumps” is still unclear, but it is probably due to an impurity-related complex; the precursor material contains a large number of residual contaminants as mentioned before (see “Structural Characterisation of the Materials Before and After the Sintering” section), some of which are present at significant concentrations (~ 100 ppm). The zero-phonon emission line of NVN at ~ 503 nm wavelength is barely detectable and cannot be distinguished from the two small shoulders (at 500 and 505 nm) in the same region. By comparing the D19 and DP 0–0.05 spectra (Fig. 9a), one can see that they superimpose well for λ > 750 nm. However, the spectra differ significantly in the 480–650 nm range due to the appearance after sintering of NVN centres, which were not present in the precursor material. In order to verify our interpretation of the main optical emission of D19 (in the range 500–650 nm) as originating from NVN centres, we compared the PL spectrum of D19 with the PL spectra of two reference samples (HPHT diamonds, Columbus NanoWorks Inc., US) of two very different sizes and both known to contain NVNs (Fig. 9b). The D19 PL spectrum (dashed line) coincides very well with those of the PL spectra of the HPHT microdiamonds containing NVNs. The emission spectrum of the 100-µm sized reference sample shows a sharp single-phonon diamond Raman line (487.4 nm) and the zero-phonon line (504 nm) of NVN under 457 nm laser excitation at room temperature (Fig. 9b, violet spectrum). The 150-nm sized HPHT nanodiamonds were excited at 488 nm, and it displayed a very similar global photoluminescence spectrum shape as that of the 100-µm sized sample, with a Raman single-phonon line at 522 nm. However, it did not exhibit the NVN zero-phonon line, behaving in that sense exactly like the D19 sample. The “bumps” present in the D19 sample spectrum are absent from both reference PL spectra, indicating that these features are not related to the NVN emission.

PL spectrum of submicron powder D19 sample at T = 293 K (blue line) compared with that of its DP 0–0.05 polycrystalline precursor (red line) under the same conditions with laser excitation at λ = 488 nm (a ), emission spectra, under 457 nm and 488 nm excitation, of two reference synthetic HPHT samples (size ~ 100 μm and < 150 nm) containing NVN centres (b ) and the Raman spectrum of D19 sample recorded using the 457 nm excitation laser radiation (c ). Arrows in (b ):lines at 487.4 nm and 522 nm are single-phonon diamond Raman lines at 457 nm and 488 nm excitation, respectively, and the line at 504 nm is the ZPL of the NVN centres. In (b):dashed line—PL spectrum of D19 at λ = 488 nm excitation (for comparison). In (c ):the diamond Raman line is centred at 1331.4 cm −1 . δ = 7.3 cm −1 is a FWHM of diamond Raman line having the Lorentzian shape
We also measured Raman scattering from D19, in the range 1000–1600 cm −1 , under excitation by 457 nm laser radiation. Figure 9c displays the Raman spectrum, corrected to remove the autofluorescence background. The spectrum consists primarily of a narrow characteristic diamond Raman line centred at 1331.4 cm −1 and an exceptionally broad (width ≈ 100 cm −1 ) band centred at 1450 cm −1 . The latter could be due to non-diamond amorphous carbon phase and/or some transpolyacetylene (TPA) species located at diamond crystals surface [1,2,3]. A further, ill-defined, band at ~ 1090 cm −1 of lower intensity is probably related to TPA species. The broad band at ~ 1450–1480 cm −1 could also be related to multivacancy chains in the diamond lattice and sp 2 - rehybridisation within these chains [6]. Footnote 7 Furthermore, we did not observe the characteristic G-band (centred at 1570–1590 cm −1 ) associated with an sp 2 graphitic nanophase. These observations are indicative that the diamond sample D19 being graphite free, which is also in agreement with its white colour under daylight illumination.
Moreover, the width of the Raman diamond line (7.3 cm −1 ) in the D19 sample is smaller than that for the DP 0–0.05 polycrystalline particles (10.6 cm −1 ). Table 1 contains Raman diamond line data for bead-milled synthetic Ib HPHT diamonds with mean size varying from 25 to 1000 nm (Microdiamant AG, Switzerland). The linewidth decreases from 9.12 to 5.24 cm −1 with increasing size. This can be explained by the lower prevalence of structural defects in larger crystals. Using data as a calibration curve to infer the crystal size from the diamond Raman linewidth of δ ≈ 7.3 cm −1 yields an estimation for the D19 crystal size of ~ 80 nm. This value coincides reasonably with the estimation in “EPR of Polycrystals and Diamond Crystallites Before and After the Sintering” section on the basis of EPR data. Moreover, this value is very similar to our previous published results where diamonds were obtained by HPHT sintering of 5-nm DND in the presence of ethanol [31]. However, the precursor material used for sintered DND and the one used in this work using the smallest (~ 25 nm) fraction of milled Du Pont shock-wave polycrystalline diamonds are considerably different from the viewpoint of crystal types and elementary crystallite sizes. This obtained size is about 8 times larger than the coherent scattering region length of L CSR ≈ 11 nm extracted from XRD earlier, but it is consistent with crystallite having a low density of structural defects, in agreement with the EPR studies.
We also studied the fluorescence from very fine individual D19 particles. For this purpose, the supernatant fraction of diluted and ultrasonicated water suspension of D19 particles obtained after centrifugation at 4500 × g for a 30 min was used. Coarse particles and large loose aggregates with size exceeding ~ 0.2–0.3 micron were absent in such supernatant. Fine D19 particles were spin-coated onto a thin glass coverslip from the diluted supernatant of D19 particles. A typical image of fine fluorescent D19 particles is shown in Fig. 10a in greyscale (mono 14-bit images). It consists of many spots with different brightness. Some spots, such as that marked by the yellow circle, have sizes close to the diffraction limit. The intensity profile of this spot has a Gaussian shape in its central core and a FWHM of about 5–6 pixel corresponding to ~ 440 nm (Fig. 10b). Such spots come from at least quarter-micron particles and particles of smaller size. A greater number of brighter spots correspond to the larger reassembled aggregates of D19 particles having more NVN colour centres and hence the overall emission intensity increasing.

Wide-field fluorescent image of isolated D19 particles obtained by confocal epifluorescence microscopy (a ), and the intensity profile of the selected nanoparticle marked by the yellow circle (b ). Image size: ~ 80 × 80 mμ. One pixel corresponds to 80 nm
Discussion
We showed that under HPHT conditions, and in the presence of ethanol, we can convert polycrystalline diamond particles (composed of tightly cemented nanometre-sized cubic diamond crystallites separated by a non-cubic diamond phase) into larger cubic diamond crystallites. The process probably occurs through recrystallisation of the cubic diamond phase and transformation of non-cubic diamond phases including multiple twin boundaries into diamond. During this process, vacancies appear and can form NVN complexes with nitrogen atom pairs. These complexes have a characteristic photoluminescence in the green. While the EPR spectra of the precursor polycrystalline diamonds show NV − triplet centres, triplet multivacancies, and SiV − centres, none of these were present after the sintering. The disappearance of NV − centres and multivacancies has previously been observed [31] after sintering detonation nanodiamonds at HPHT conditions (P = 7 GPa and T ≥ 1350 °C) with ethanol. Footnote 8 The presence of multivacancies is a characteristic feature of damaged diamond lattices, with defects mainly located in a thin layer of ~ 2 nm at the surface. The absence of the paramagnetic triplet and SiV colour centres is strong evidence that substantial recrystallisation took place, accompanied with the appearance of new defect types (NVN). The saturation trend of the substitutional nitrogen (P1 centre) EPR signal with increasing microwave power indicates long spin–spin and spin–lattice relaxation times. These are signatures of improvement of the quality of diamond crystal lattice after sintering. We can assume that during HPHT sintering, vacancies from empty spaces within or between polycrystals join with A-centres to form the new NVN entities.
Conclusiones
Sintering of diamond polycrystals, with size varying from 3 to 50 nm, in the presence of ethanol, lead to the substantial enlargement of elementary diamond nanocrystals and improved their crystalline quality. During this process, SiV − and NV − colour centres present in the precursor nanodiamond disappeared, while the EPR signature of P1 substitutional nitrogen paramagnetic centres appeared. We also observed the green photoluminescence of NVN colour centres. The comparison of the FWHM of diamond Raman line (~ 1332 cm −1 ) of the synthesised selected microcrystals under study with those of a series of reference samples revealed that the mean size of diamond crystals after sintering is approximately 80 nm. The analysis of the EPR spectrum dependence upon microwave power demonstrated the good crystalline quality of the synthesised sintered diamond with a concentration of P1 centres smaller than 200 ppm. Hence, our technique of HPHT sintering is a strong alternative to conventional high-energy particle beam irradiation [9] to form NVN centres in nanodiamond. It can be used to produce purely “green” fluorescing nanodiamonds with no (or very limited) crosstalk with the “red” fluorescing nanodiamonds (containing NV 0 and NV − centres), as required in biolabelling for cathodoluminescence integrated correlation electron-light microscopy [32].
Disponibilidad de datos y materiales
The data underpinning this manuscript is available from the corresponding author on request.
Notes
- 1.
The apparatus response function of the XRD diffractometer was determined from a LaB6 reference sample.
- 2.
One mechanism explaining the lack of HFS in the EPR signal of paramagnetic nitrogen in size < 50 nm diamond nanoparticles is described in detail elsewhere [21]. Alternative mechanisms are also not excluded [33, 34].
- 3.
X-ray diffraction CSR size is ~ 4 times smaller in this case.
- 4.
This narrow EPR signal is slightly asymmetrical and consists of at least two components related to P1 centres and to defects having C–C dangling bond spins S = 1/2. The intensity of the second component represents at least 30% of the intensity of the main peak.
- 5.
Here we assume the sequence of untreated first-derivative experimental EPR spectra measured at different P MW , two of which are shown in Fig. 7a.
- 6.
However, the Raman line was detected at 457 nm excitation.
- 7.
The Raman band centred at ~ 1480 cm −1 was found for HPHT diamonds grown with magnesium-based catalysts [6]. Such diamond crystals may contain SiV and GeV colour centres.
- 8.
Instead of a multivacancy g = 4.00 singlet EPR signal, a new EPR signal with a quintet hyperfine structure related to N⋯N pairs separated by no more than 0.7 nm was observed in this case.
Abreviaturas
- NV − :
-
Vacante de nitrógeno
- NVN:
-
Nitrogen-vacancy-nitrogen
- HPHT:
-
High-pressure, high-temperature
- DND:
-
Detonation nanodiamond
- EPR:
-
Electron paramagnetic resonance
- FWHM:
-
Ancho completo a la mitad del máximo
- XRD:
-
Difracción de rayos X
- LCSR :
-
Coherent scattering region length
- HRTEM:
-
Microscopía electrónica de transmisión de alta resolución
- PL:
-
Fotoluminiscencia
- MW:
-
Microwave
- HFS:
-
Hyper-fine structure
- RT:
-
Temperatura ambiente
Nanomateriales
- ¿Qué es el torneado de diamantes?
- ¿Qué es la sinterización selectiva por láser?
- Diamante
- Nanodiamantes para sensores magnéticos
- El efecto de una pequeña cantidad de SiO2 en la cinética de sinterización de nanopolvos tetragonales de zirconia
- Modelado y simulación de dinámica molecular del corte con diamante de cerio
- Estrategia de sinterización asistida por hidrotermia hacia material de ánodo LiNb3O8 de estructura hueca y porosa
- Matrices de nanopilares de GaAs con tapa de Au fabricadas mediante grabado químico asistido por metal
- Comportamiento de sinterización de SiC sinterizado por plasma de chispa con nanopartículas compuestas de Si-SiC preparadas mediante el proceso de plasma térmico de CC
- Células solares de perovskita fabricadas con un aditivo polar aprótico respetuoso con el medio ambiente de 1,3-dimetil-2-imidazolidinona
- Propiedades termoeléctricas del SnSe policristalino bidopado tipo n prensado en caliente