


Bandgap ajustable por deformación y alta movilidad del portador en monocapas de SiAs y SiAs2 de los estudios de los primeros principios
Resumen
La búsqueda de nuevos materiales bidimensionales (2D) delgados atómicamente independientes estables es de gran interés en los aspectos fundamentales y prácticos de las ciencias de los materiales contemporáneos. Recientemente, se ha realizado la síntesis de monocristales de SiAs estratificados, lo que indica que su estructura de pocas capas puede exfoliarse mecánicamente. Realizando cálculos de teoría funcional de densidad de primeros principios, propusimos dos SiAs y SiAs semiconductores dinámicamente y termodinámicamente estables 2 monocapas. El cálculo de la estructura de la banda revela que ambos exhiben espacios de banda indirectos y una transición de banda indirecta a directa incluso al metal se encuentran mediante la aplicación de tensión. Además, encontramos que SiAs y SiAs 2 las monocapas poseen una movilidad de portadora mucho mayor que MoS 2 y muestran transporte anisotrópico como el fosforeno negro, lo que los convierte en una aplicación potencial en optoelectrónica. Nuestros trabajos allanan una nueva ruta a nanoescala para funcionalidades novedosas de dispositivos ópticos.
Antecedentes
Los cristales bidimensionales (2D) atómicamente delgados se han convertido en uno de los campos de mayor crecimiento de la ciencia de los materiales contemporánea. Las propiedades electrónicas versátiles, la excelente movilidad de los electrones y las aplicaciones prometedoras en nanoelectrónica y optoelectrónica están impulsando a un gran porcentaje de físicos de materia condensada a buscar nuevos materiales 2D. Después del grafeno [1–4], se sintetizó una gran cantidad de otros materiales 2D, como el siliceno [5–7], nanohojas de nitruro de boro [8, 9], dicalcogenuros de metales de transición (TMD) [10, 11], fósforo negro [12, 13], borofeno [14-16], arseneno [17, 18], telureno [19] y sus compuestos isoelectrónicos [20-23]. La lista de materiales 2D se está expandiendo rápidamente, y ahora se conocen más de miles de tipos de tales materiales, que abarcan el espectro completo de propiedades electrónicas y de otro tipo. Y sus propiedades novedosas, diferentes o incluso mejores que las de sus homólogos a granel, se predicen teóricamente y se confirman experimentalmente con firmeza.
Aunque se invirtieron grandes y sustanciales esfuerzos para encontrar diversos materiales 2D, incluidos algunos que ya poseen bandgaps u otras propiedades deseables, no se ha llegado a un consenso. El grafeno con una maravillosa movilidad de portador, alta estabilidad mecánica y electrones dirac sin masa ha atraído mucha atención hasta la fecha, pero la falta de una banda prohibida intrínseca dificulta su aplicación en la industria de dispositivos electrónicos modernos. Aunque se han realizado grandes esfuerzos, no se ha logrado abrir una brecha de banda considerable sin efectos secundarios [24, 25]. Los TMD con alto rendimiento en dispositivos optoelectrónicos tienen de hecho una banda prohibida intrínseca, pero exhiben poca movilidad de portadora [26-28]. El fósforo negro y azul con una banda prohibida sintonizable sensible a la deformación y una alta movilidad del portador anisotrópico no puede mantenerse estable en el aire [13, 29]. Recientemente, la síntesis de capas de SiAs y SiAs 2 Se han realizado monocristales [30-32], lo que indica que se pueden obtener pocas estructuras de capas mediante exfoliación mecánica.
En el presente trabajo, basado en cálculos de teoría funcional de densidad (DFT) de primeros principios, propusimos dos monocapas semiconductoras dinámicamente y termodinámicamente estables SiAs y SiAs 2 . Ambos poseen intervalos de banda indirectos (2,39 eV y 2,13 eV respectivamente). La aplicación de deformación isotrópica a lo largo de dos direcciones en el plano prácticamente transforma los SiAs (SiAs 2 ) monocapa en un material de separación directa de 1,75 eV (1,60 eV). Además, encontramos que SiAs y SiAs 2 las monocapas poseen una movilidad de portadora mucho mayor que MoS 2 y muestran transporte anisotrópico como el fosforeno negro, lo que los convierte en una aplicación potencial en optoelectrónica. Nuestros trabajos allanan una nueva ruta a nanoescala para funcionalidades novedosas de dispositivos ópticos.
Métodos computacionales
Los cálculos de DFT se realizan utilizando el código del paquete de simulación ab initio de Viena (VASP) [33]. Se utilizó la función de intercambio-correlación de Perdew-Burke-Ernzerhof (PBE) [34] bajo la aproximación de gradiente generalizado (GGA). Se empleó el método de onda aumentada del proyector (PAW) [35] para describir la interacción electrón-ión. Se aplicó un vacío de 20 Å perpendicular a las láminas (a lo largo del eje c) para evitar la interacción entre capas. Se utiliza un corte de energía cinética de 500 eV para el conjunto básico de ondas planas. El muestreo de la zona de Brillouin se realiza con una cuadrícula de Monkhorst-Pack [36] de 15 × 5 × 1 para láminas 2D. Los criterios de convergencia empleados tanto para la relajación electrónica autoconsistente como para la relajación iónica se establecen en 10 −4 y 0,01 eV / Å para energía y fuerza, respectivamente. Los cálculos de fonones se llevan a cabo utilizando el método de supercélulas a través del código PHONOPY [37, 38], y las constantes de fuerza en el espacio real de las supercélulas se calculan en la teoría de perturbación funcional de densidad (DFPT) implementada en VASP. Además, una energía más estricta (10 −8 eV / átomo) y criterio de convergencia de fuerzas (10 −4 eV / Å) se utilizan durante los cálculos de espectros vibracionales. En los cálculos de dinámica molecular (MD), se emplean supercélulas (3 × 3 × 1) y la temperatura se mantiene a 300 K durante 6 ps con un intervalo de tiempo de 2 fs en el conjunto moles-volumen-temperatura (NVT). Los espectros raman se calcularon al nivel de teoría PBE utilizando el código CASTEP [39-41].
Resultados y discusiones
Las estructuras geométricas y la distribución de la densidad electrónica de SiAs y SiAs 2D independientes y relajados 2 se presentan en la Fig. 1a, b, respectivamente, y sus estructuras a granel se muestran en el archivo adicional 1:Figura S1 del material suplementario. Como se muestra en el archivo adicional 1:Figura S1a yb, los SiAs a granel (SiAs 2 ) posee simetría C2 / m (Pbam) y consta de capas de Si-As apiladas débilmente unidas por fuerzas de van der Waals con una distancia de 3,06 Å (1,66 Å). La celda unitaria de SiAs monocapa es rómbica y sus parámetros de cristal optimizados son a 1 =3.69Å y b 1 =10,83Å con φ =99,81 °. SiAs contiene 6 átomos de Si y 6 átomos de As. Cada átomo de Si tiene cuatro átomos vecinos más cercanos (3 As y 1 Si), mientras que cada átomo de As forma sólo tres enlaces covalentes con átomos de Si vecinos. Existen dos tipos de enlaces, a saber, enlaces Si-Si y Si-As. Y la longitud del enlace Si-Si es de aproximadamente 2,35 Å y la de Si-As está en el rango de 2,39 Å y 2,43 Å, y la altura de pandeo es d 1 =4,86 Å. En la vista lateral de SiAs monocapa, se forma una estructura similar a un cordón de gafas con capas dobles y simples abultadas alternativamente. Otra estructura monocapa de compuesto de silicio y arsénico es SiAs 2 . Su celda principal contiene 4 átomos de Si y 8 átomos de As, con una estructura rectangular y los parámetros de cristal optimizados son a 2 =3.68Å y b 2 =10,57 Å. Cada átomo de As tiene tres átomos de Si vecinos más cercanos o forma un enlace covalente con átomos de Si vecinos y dos enlaces covalentes con ellos mismos, mientras que cada átomo de Si tiene sólo cuatro átomos de As vecinos más cercanos. A diferencia del primero, SiAs 2 posee un enlace As-As más débil (2,50 Å) en lugar de un enlace Si-Si. Y sus enlaces Si-As varían de 2,41 Å a 2,45 Å, y la altura de pandeo es d 2 =5,09 Å. A partir de la distribución de la densidad de electrones, los átomos de As atraen los electrones de los átomos de Si por su gran electronegatividad y tienen una mayor densidad de electrones. Con el fin de ayudar a la caracterización experimental futura, calculamos y verificamos los espectros Raman de SiAs y SiAs a granel y monocapa 2 . Se han observado cambios claros entre la monocapa y los cristales completos en el archivo adicional 1:Figura S2 del material complementario, cuyos orígenes se han identificado como la influencia de la interacción de las capas de van der Waals [42].
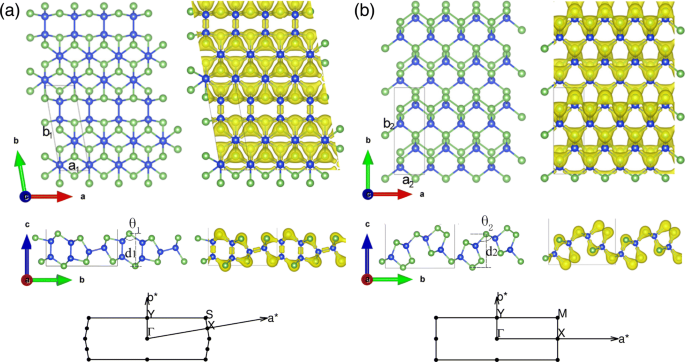
Estructura geométrica y distribución de la densidad electrónica de las monocapas de SiAs y SiAs 2 . (Color en línea) Vistas superior y lateral de monocapas a SiAs y b SiAs 2 estructura geométrica y distribución de densidad de electrones y la zona de Brillouin asociada. La bola azul y verde indican el átomo de Si y As, respectivamente
Para conocer la estabilidad de SiAs (SiAs 2 ), primero calculamos la energía cohesiva, definida como E coh =( nE Si + mE Como - E Mono ) / ( n + m ), donde E Si , E Como y E Mono son las energías totales de un solo átomo de Si, un solo átomo de As y una unidad de fórmula de SiAs monocapa (SiAs 2 ), respectivamente, y n (m) es el número de átomos de As (Si) en la unidad de fórmula. Nuestros cálculos muestran que la monocapa de SiAs tiene una energía cohesiva de 5,13 eV / átomo, que es un poco más grande que la de SiAs 2 monocapa 4,98 eV / átomo. A modo de comparación, al mismo nivel teórico, las energías cohesivas del arseneno y el siliceno son 2,99 y 3,71 eV / átomo, respectivamente [18, 43]. Las altas energías cohesivas de SiAs y SiAs 2 revelan que ambos están fuertemente unidos con alta estabilidad.
Para confirmar aún más las estabilidades estructurales de SiAs monocapa y SiAs 2 , también hemos realizado cálculos de espectros de fonones vibracionales. Como se muestra en la Fig. 2a, las frecuencias positivas representan la mayoría de los modos excepto el modo acústico transversal cerca del Γ point, que se debe al ablandamiento de los fonones y se ha informado en otros sistemas similares [44, 45], lo que indica que las estructuras son dinámicamente estables. Luego, llevamos a cabo simulaciones de MD de los primeros principios de 6 ps a temperatura ambiente ( T =300 K ), como se presenta en la Fig. 2b. La ligera fluctuación de energía y las estructuras bien mantenidas sugieren que son térmicamente estables a temperatura ambiente. Nuestros resultados implican que las monocapas SiAs y SiAs 2 podría realizarse experimentalmente a temperatura ambiente.
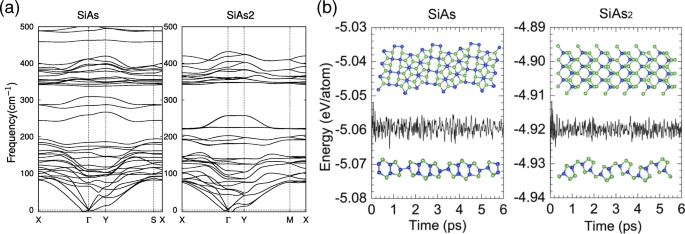
Curvas de dispersión de fonones y simulaciones MD de monocapas SiAs y SiAs 2 . un Las curvas de dispersión de fonones para SiAs monocapa y SiAs 2 . b Relaciones de energía total y tiempo durante simulaciones MD a temperatura ambiente de SiAs y SiAs 2 . También se proporcionan instantáneas seleccionadas de las estructuras monocapa al final de 6 ps
Con las estructuras optimizadas de SiAs monocapa y SiAs 2 , ahora prestamos atención a sus propiedades electrónicas. Las estructuras de bandas de descomposición orbital calculadas de SiAs y SiAs 2 Las monocapas se muestran en la Fig. 3. Nuestros cálculos muestran claramente que SiAs y SiAs 2 las monocapas son semiconductores indirectos con amplios intervalos de banda. Para SiAs monocapa, el máximo de la banda de valencia (VBM) se encuentra en la Y punto, mientras que el mínimo de banda de conducción (CBM) está en el Γ (Figura 3a). La banda prohibida indirecta de SiAs monocapa es E g =1,72 eV dentro del esquema PBE. También se puede ver que el estado de VBM en Y el punto comprende la p y orbital, mientras que el CBM de Γ El punto comprende principalmente el orbital s, lo que significa que la deformación externa tendrá efectos diferentes en los dos estados y puede conducir a una transición indirecta-directa, como se revela a continuación. A diferencia de SiAs, la monocapa SiAs 2 es un semiconductor casi directo con VBM ubicado al lado de la Y punto y CBM está un poco desplazado de él (Fig. 3b). Los SiAs 2 la banda prohibida indirecta monocapa es E g =1,42 eV dentro del esquema PBE. Y el VBM y CBM de SiAs 2 monocapa se componen de la p y orbital y s orbital, respectivamente. Para obtener un valor de banda prohibida más preciso, también realizamos los cálculos funcionales híbridos (HSE06) [46, 47] para SiAs y SiAs 2 monocapas. A partir de las estructuras de banda calculadas (la parte derecha de la Fig.3a, b), los elementos punzantes de los estados de banda de PBE y HSE son básicamente los mismos, y la banda prohibida indirecta todavía se predice dentro de los cálculos funcionales híbridos, pero el valor de la brecha es aumentado a 2,39 eV y 2,07 eV para SiAs y SiAs 2 , respectivamente.
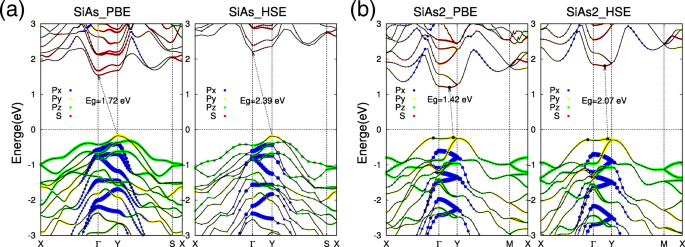
Estructuras de bandas de SiAs y SiAs monocapa 2 calculado por PBE y HSE06. La descomposición orbital electrónica de estructuras de bandas de monocapas SiAs y SiAs 2 se representan como a y b , respectivamente. Los puntos rojos denotan s orbital, mientras que azul, amarillo y verde son p x , p y y p z , respectivamente. El nivel de Fermi se establece en cero y se indica con una línea de puntos
La movilidad del portador, que es un factor clave de las aplicaciones potenciales en los dispositivos electrónicos modernos para los materiales 2D recién descubiertos, es tan importante como la banda prohibida y la ubicación de CBM y VBM. Para obtener más detalles sobre las propiedades de la estructura electrónica de SiAs y SiAs 2 monocapas, luego calculamos sus movilidades portadoras limitadas por fonones acústicos (incluidos el electrón y el agujero en las direcciones xey) sobre la base de la teoría del potencial de deformación (DP) [48] a temperatura ambiente ( T =300 K ). En el régimen de baja energía (300 K ), la dispersión de electrones-fonones acústicos domina el transporte de portadores, lo que hace que el fonón acústico limitado sea una forma eficaz de predecir las movilidades de portadores de muchas estructuras 2D, como MoS 2 monocapa [49], telureno [19], fosfeno [50] y pocas capas de MoO 3 [51]. Las masas efectivas calculadas m ∗ y movilidades de portadores μ de SiAs y SiAs 2 las monocapas muestran que ambos son de alta movilidad y anisotropía de transporte (consulte el archivo adicional 1:Tabla S1 y las Figuras S3 y S4) como el fosforeno negro [50]. Estimar la movilidad del portador de SiAs y SiAs 2 , en primer lugar realizamos un ajuste de sus bandas utilizando el modelo de electrones casi libres para obtener las masas portadoras efectivas. Para SiA, definimos x y y como la dirección perpendicular a los vectores de celosía b y a , respectivamente. \ (M_ {e} ^ {*} \) y \ (m_ {h} ^ {*} \) a lo largo de la dirección x son aproximadamente 0,15 m 0 y 0,86 m 0 , respectivamente, ya lo largo de la dirección y son 0,80 m 0 y 0,22 m 0 ( m 0 es la masa de electrones libres), respectivamente. Para SiAs 2 , la dirección del vector de celosía a se define como x , mientras que el de b es y . \ (M_ {e} ^ {*} \) y \ (m_ {h} ^ {*} \) a lo largo de la dirección x son aproximadamente 0,14 m 0 y 0,65 m 0 , respectivamente, ya lo largo de la dirección y son 2,05 m 0 y 1,82 m 0 , respectivamente. Además, estudiamos las constantes elásticas (C) y los potenciales de deformación (E1) (consulte el archivo adicional 1:Figura S2 y S3). Basado en el m obtenido anteriormente ∗ , C y E1, estimamos la movilidad del portador como se indica en la Tabla 1. Las movilidades de electrones para SiAs (SiAs 2 ) a lo largo de x y y las direcciones son 0,66 (0,26) y 0,54 (0,11) × 10 3 · cm 2 V −1 S −1 , mientras que la movilidad del agujero a lo largo de x y y las direcciones son 3,90 (0,13) y 0,30 (0,65) × 10 3 · cm 2 V −1 S −1 , respectivamente, ambos muy superiores a los de MoS 2 [49].
Para arrojar más luz sobre el mecanismo de enlace subyacente de los átomos de Si y As en las monocapas de SiAs y SiAs 2 , la densidad total y parcial de estados (PDOS) de ellos usando PBE funcional, con su distribución de densidad electrónica correspondiente a VBM y CBM, se proporcionan en la Fig. 4, respectivamente. Se puede ver que el PDOS de los átomos de As y Si (Fig. 4a, c) muestra una fuerte hibridación de s y p orbitales, lo que indica el fuerte enlace covalente entre ellos. Las distinciones entre monocapas SiAs y SiAs 2 son la localización de p z orbital, que se atribuyen a los diferentes entornos de coordinación de enlace del átomo de As. Los estados de electrones del par solitario, localizados en el átomo de As tanto en SiAs como en SiAs 2 monocapas, aumente los tres orbitales de enlace más cercanos para decidir la formación de pandeo de la estructura de la monocapa y para formar la p z Acción de localización orbital. En SiAs monocapa, los pares solitarios están separados por el enlace Si-As, que relaja el efecto repulsivo y amplía la p z orbital. Mientras que en la monocapa SiAs 2 , As-As bond, siendo la situación que es muy común en los semiconductores del grupo V, localiza la p z orbital en un nivel de energía más profundo.
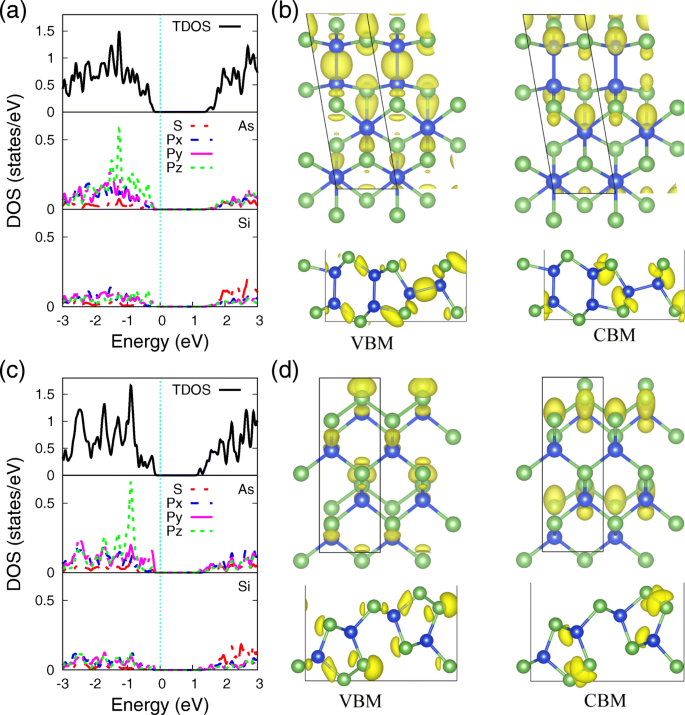
Densidad proyectada de estados y densidad electrónica de VBM y CBM. La densidad proyectada de estados (PDOS) de los átomos de As y Si y la distribución de la densidad electrónica correspondiente a VBM y CBM de ( a , b ) SiAs y ( c , d ) SiAs 2 monocapas. El valor de isosuperficie 0.034 e / Å 3
Como sabemos, el carácter de los estados de frontera no solo es de interés para una comprensión microscópica de los canales de conducción, sino también de gran interés para el diseño de contactos óptimos. [52] Las densidades de carga correspondientes a VBM y CBM de monocapas SiAs y SiAs 2 se presentan en la Fig. 4b yd, respectivamente. El VBM es casi la hibridación de orbitales 3p de Si y As, mientras que CBM es principalmente de la contribución de los orbitales 3s de Si y As, que también son consistentes con los resultados de PDOS en la Fig.4a, cy la descomposición orbital electrónica de estructuras de bandas en la Fig. 3.
La deformación mecánica es una forma eficaz de modular las propiedades electrónicas de los materiales 2D, que se utilizan ampliamente para modificar la estructura de bandas de los fosfornos negros y azules y otros materiales de nanoplacas [53-55]. Especialmente, para el sistema de estructura pandeada, el costo de energía suele ser bastante pequeño para inducir una tensión marcada. Aquí, la aplicación de deformación mecánica se simula variando la constante de la red, así como los grados internos de libertad de cada átomo durante la optimización geométrica. La cepa ε se define como ε =( l - l 0 ) / l 0 , donde l y l 0 son las constantes de celosía tensas y de equilibrio de las monocapas SiAs y SiAs 2 . En la Fig. 5a, b, las variaciones detalladas de la estructura geométrica de alto pandeo de SiAs 2D y SiAs 2 bajo tensiones están representadas, respectivamente. Se puede ver que sus alturas de pandeo se expanden o comprimen al cambiar el ángulo de pandeo θ 1 (2) con deformaciones biaxiales de compresión o tracción en variaciones casi lineales. Y también encontramos que su estructura geométrica de alto pandeo se mantienen bien bajo tensiones bastante grandes, cuyos espectros de fonones, como se muestra en el archivo adicional 1:Figura S5 y S6, no existen frecuencias negativas incluso en el régimen de gran tensión. Las variaciones de espacios de SiAs monocapa y SiAs 2 bajo deformaciones biaxiales de compresión y tracción se muestran en las Fig. 5c, d, respectivamente. Se puede ver que las propiedades electrónicas de SiAs y SiAs 2 dependen sensiblemente de la deformación y se someten a una transición de banda indirecta a directa en cierta región de deformación y luego al metal en una región de deformación grande.
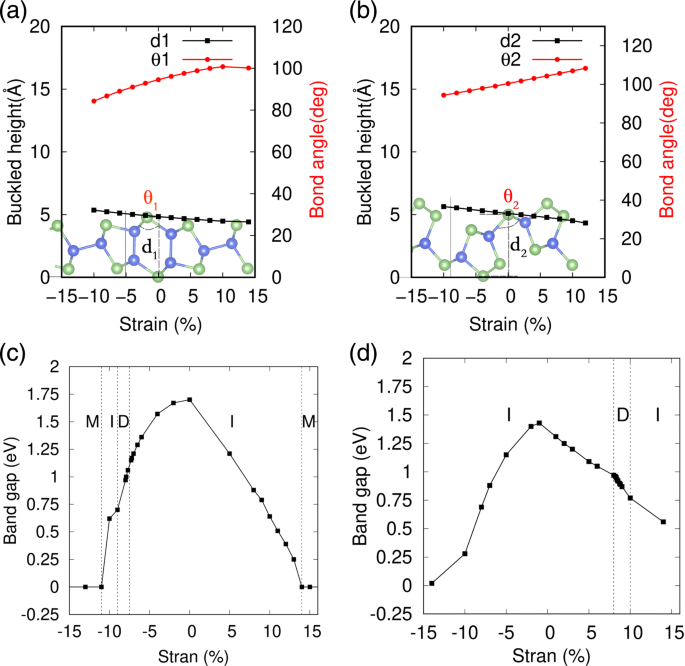
Efectos de deformación en las estructuras geométricas y los espacios de banda de SiAs y SiAs 2D 2 . un , c representan SiAs; y b , d denotar SiAs 2 ; M, I y D representan metal, semiconductor indirecto y semiconductor directo, respectivamente
Las variaciones detalladas de SiAs y SiAs 2 las estructuras de bandas se muestran en las Figs. 6 y 7, respectivamente. Bajo tensiones compresivas biaxiales, la altura de pandeo de SiAs monocapa está aumentando y el CBM cambia de Γ hasta un punto en la línea Y – S y de regreso a Y. Mientras que el VBM se mantiene quieto en el punto Y hasta que la deformación por compresión alcanza ε =- 10 % . Por lo tanto, al aumentar la deformación por compresión, la banda prohibida cambia de Y indirecta a Γ , a través de Y indirecta a un punto en la línea Y – S, para dirigir Y a Y y de regreso a un punto indirecto en la Γ –Y línea a Y, como se muestra en la Fig. 6. Para deformaciones por tracción, el VBM en Y se mueve a un punto en la línea Y – S y el CBM en Γ se mueve a Y y la banda prohibida permanece indirecta. Para grandes deformaciones, no importa si la compresión o la tracción conducen a una transición de metal, como se muestra en la Fig. 5c.
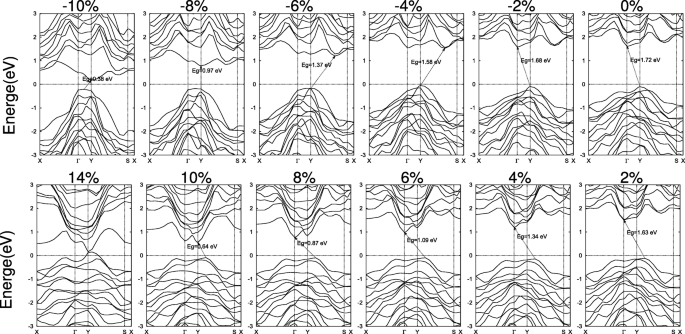
Estructuras de bandas de SiAs 2D bajo las deformaciones biaxiales. El nivel de Fermi se establece en cero y se indica con una línea de puntos
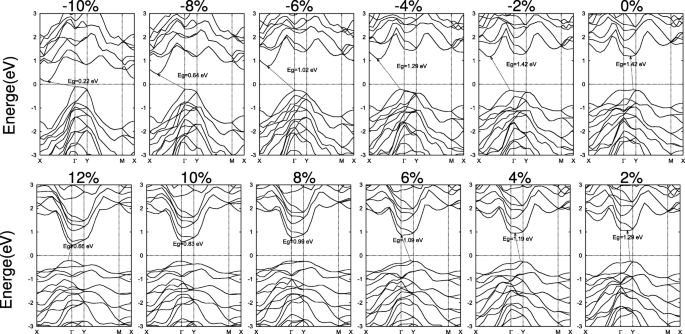
Estructuras de bandas de SiAs 2D 2 bajo las tensiones biaxiales. El nivel de Fermi se establece en cero y se indica con una línea de puntos
En la figura 7, se ha realizado un estudio similar para 2D SiAs 2 . En lugar de compresión, las deformaciones por tracción en el rango de 8 a 10% dan como resultado espacios de banda directos. cuando la monocapa SiAs 2 se extiende con una disminución de la altura de pandeo bajo las tensiones de tracción, el desplazamiento de VBM desde un punto en el Γ –Y línea a Γ y mantenerse quieto en el rango de 8-10% y luego cambiar a un punto en el Γ –X línea, mientras que el CBM se mueve desde un punto en la Γ –Y línea a Γ y espera. Por lo tanto, con el aumento de la deformación por tracción, la banda prohibida cambia de indirecta en el Γ –Y línea para dirigir Γ - Γ y luego de vuelta a un punto indirecto en el Γ –X línea a Γ , como se ilustra en la Fig. 7. Las deformaciones por compresión siguen siendo la banda prohibida indirecta. Y las cepas grandes tienen efectos similares, lo que lleva a una transición de metal como SiAs.
Estructuras de bandas directas representativas de SiAs y SiAs deformados 2 también se muestran en el archivo adicional 1:Figura S7a yb por los cálculos de PBE y HSE. Para SiA, una banda prohibida directa de E g =1,75 eV (HSE) con VBM y CBM localizados en Y puntos se obtiene bajo una tensión de compresión biaxial de ε =- 7.5 % . A diferencia de los SiAs, una deformación por tracción biaxial de ε =8.5 % induce el SiAs 2 a una banda directa de E g =1,60 eV (HSE). Y el VBM y CBM están en el Γ punto.
Conclusiones
En resumen, realizando un cálculo de DFT de primeros principios, hemos propuesto dos nuevos tipos de materiales 2D de compuestos de silicio y arsénico, SiAs y SiAs 2 , que son tanto dinámica como termodinámicamente estables. Nuestros cálculos muestran que SiAs y SiAs 2 las monocapas son semiconductores indirectos con intervalos de banda de 2,39 eV y 2.07 eV , respectivamente. La banda prohibida de SiAs y SiAs 2 Las monocapas son sensibles a la deformación, que experimentan una transición de banda indirecta a directa e incluso al metal ante cierta deformación mecánica. SiAs y SiAs 2 las monocapas poseen una mayor movilidad que MoS 2 y muestran transporte anisotrópico como el fosforeno negro. Nuestros trabajos allanan una nueva ruta a nanoescala para nuevas funcionalidades de dispositivos ópticos.
Abreviaturas
- 2D:
-
Bidimensional
- CASTEP:
-
Paquete de energía total secuencial de Cambridge
- CBM:
-
Banda de conducción mínima
- DFT:
-
Teoría funcional de la densidad
- DFPT:
-
Teoría de la perturbación funcional de la densidad
- DP:
-
Potencial de deformación
- GGA:
-
Aproximación de gradiente generalizada
- MD:
-
Dinámica molecular
- NVT:
-
Moles-volumen-temperatura
- PAW:
-
Proyector de onda aumentada
- PBE:
-
Perdew-Burke-Ernzerhof
- PDOS:
-
Densidad parcial de estados
- TMD:
-
Dicalcogenuros de Transiton-metal
- VASP:
-
Paquete de simulación Viena ab initio
- VBM:
-
Máximo de banda de valencia
Nanomateriales
- Eliminación por adsorción de iones de cobre (II) de una solución acuosa utilizando un nano-adsorbente de magnetita de residuos de cascarilla de molino:síntesis, caracterización, adsorción y mode…
- Absorbedor perfecto de banda ultra estrecha y su aplicación como sensor plasmónico en la región visible
- Estudio de los primeros principios sobre la estabilidad y la imagen STM del borofeno
- RGO y redes de grafeno tridimensionales co-modificadas TIM con alto rendimiento
- Preparación de nanoesferas poliméricas impresas con iones de paladio (II) y su eliminación del paladio (II) de la solución acuosa
- Nanoconjuntos de ácido 5-aminolevulínico-escualeno para fotodetección y terapia de tumores:estudios in vitro
- Estudio de los primeros principios de los defectos puntuales en la superrejilla de GaAs / AlAs:la estabilidad de fase y los efectos sobre la estructura de la banda y la movilidad del portador
- Propiedades electrónicas ajustables por deformación y alineaciones de bandas en heteroestructura GaTe / C2N:un cálculo de los primeros principios
- Investigación de la banda de energía en las heterouniones de disulfuro de molibdeno y ZrO2
- Bandgap ajustable por deformación y alta movilidad del portador en monocapas de SiAs y SiAs2 de los estudios de los primeros principios
- Preparación de micro / nanoesferas huecas de polianilina y su capacidad de eliminación de Cr (VI) de aguas residuales