


Propiedades electrónicas ajustables por deformación y alineaciones de bandas en heteroestructura GaTe / C2N:un cálculo de los primeros principios
Resumen
Recientemente, GaTe y C 2 Las monocapas N se han sintetizado con éxito y muestran fascinantes propiedades electrónicas y ópticas. Tal híbrido de GaTe con C 2 El N puede inducir nuevas propiedades físicas novedosas. En este trabajo, realizamos simulaciones ab initio sobre las propiedades estructurales, electrónicas y ópticas del GaTe / C 2 Heteroestructura de N van der Waals (vdW). Nuestros cálculos muestran que GaTe / C 2 La heteroestructura de N vdW es un semiconductor de intervalo indirecto con alineación de banda de tipo II, que facilita una separación efectiva de los portadores fotogenerados. Curiosamente, también presenta una absorción de luz UV visible mejorada en comparación con sus componentes y se puede adaptar para ser un buen fotocatalizador para la división del agua a cierto pH mediante la aplicación de deformaciones verticales. Además, exploramos específicamente la adsorción y descomposición de moléculas de agua en la superficie de C 2 Capa de N en la heteroestructura y la posterior formación de hidrógeno, que revela el mecanismo de producción de hidrógeno fotocatalítico en el 2D GaTe / C 2 N heteroestructura. Además, se ha encontrado que las deformaciones biaxiales en el plano pueden inducir transiciones indirecto-directo-indirecto, semiconductor-metal y tipo II a tipo I o tipo III. Estos interesantes resultados hacen que GaTe / C 2 La heteroestructura de N vdW es un candidato prometedor para aplicaciones en la próxima generación de dispositivos optoelectrónicos multifuncionales.
Antecedentes
Desde el descubrimiento del grafeno [1, 2], el interés por los materiales en capas bidimensionales (2D) ha ido creciendo de manera constante. Muchos materiales 2D similares al grafeno, como los dicalcogenuros de metales de transición [3], las estructuras de panal de abejas monocapa de elementos del grupo V y los compuestos binarios III-V [4-8], y los calcogenuros de metales de transición posteriores (PTMC) [9] han ganado mucho de interés por sus excepcionales propiedades físicas y sus prometedoras aplicaciones. Entre estos diversos materiales 2D, la monocapa de GaTe, como miembro de las PTMC [9], ha sido fabricada con éxito mediante epitaxia de haz molecular [10]. Los cálculos teóricos mostraron que la monocapa de GaTe es un semiconductor de banda prohibida indirecta y su banda prohibida se puede modular aplicando tensiones [11]. Además, monocapa C 2 N, un nuevo material en capas 2D con distribuciones uniformes de poros y átomos de nitrógeno, también se sintetizó con éxito mediante una reacción química húmeda ascendente y se descubrió que es un semiconductor de espacio directo [12]. Muchos estudios demostraron que su banda prohibida, las posiciones del borde de la banda y las propiedades ópticas se pueden diseñar variando su orden de apilamiento, número de capa, campo eléctrico externo o deformación y aleando / sustituyendo con otros elementos [13-16]. Cabe señalar que la banda prohibida directa sintonizable y la naturaleza porosa de C 2 Se espera que el N presente propiedades deseables para la electrónica, la optoelectrónica y la conversión de energía, así como para la división fotocatalítica del agua, etc. [15]. Sin embargo, todavía queda un desafío importante para el uso de C 2 N en fotocatálisis y células fotovoltaicas:los pares de electrones y huecos fotogenerados permanecen en las mismas regiones espacialmente, lo que puede conducir a una alta tasa de recombinación de portadores fotogenerados, reduciendo así la conversión de energía solar
Paralelamente a los esfuerzos en materiales 2D únicos, las heteroestructuras de van der Waals (vdW) fabricadas apilando diferentes materiales semiconductores 2D han abierto nuevas vías para crear nuevos materiales y diseñar nuevos dispositivos [17-23]. Este tipo de heteroestructura se puede clasificar generalmente en tres tipos:el tipo I (brecha transversal), el tipo II (brecha escalonada) y el tipo III (brecha rota) de acuerdo con las posiciones relativas del máximo de la banda de valencia (VBM) y la banda de conducción. mínimo (CBM) de los respectivos semiconductores [18, 24, 25]. Para las heteroestructuras de tipo I, las energías del VBM y CBM de un material se encuentran a caballo entre las del otro material y todos los electrones y huecos fotogenerados se acumulan en la misma capa, lo que induce la recombinación ultrarrápida de los portadores excitados y, por lo tanto, se pueden utilizar. en dispositivos optoelectrónicos, como diodos emisores de luz. En el caso de heteroestructuras de tipo II, tanto el CBM como el VBM de un material son más bajos o más altos en energía que los del otro material. Como resultado, los electrones y huecos fotogenerados se confinan por separado en los dos materiales, respectivamente, inhibiendo así la velocidad de recombinación. Por tanto, pueden utilizarse como componentes básicos para dispositivos fotovoltaicos [18, 24]. En cuanto a las heteroestructuras de tipo III, el nivel de VBM de un material es más alto que el nivel de CBM del otro, lo que es deseable para transistores de efecto de campo de efecto túnel [25, 26]. Muy recientemente, muchas heteroestructuras basadas en GaTe se han estudiado extensamente tanto teórica como experimentalmente. La heteroestructura GaTe / InSe ha sido fabricada experimentalmente y presenta la alineación de banda de tipo II [27, 28]. La heteroestructura cuasi-2D GaTe / GaSe se creó transfiriendo GaSe exfoliado de pocas capas a láminas de GaTe a granel y se encontró que formaba una alineación de banda de tipo I en la interfaz [29]. Se verificó que la heteroestructura GaTe / SnI es un aislante Hall de espín cuántico de gran espacio y exhibe una notable división de Rashba que puede modularse cambiando la distancia entre capas de las heterosheets [30]. Además, la construcción de semiconductores / C 2 N heteroestructuras, como g-C 3 N 4 / C 2 N [31], MoS 2 / C 2 N [32] y CdS / C 2 N [33], demostró un enorme potencial para promover el rendimiento fotocatalítico de C 2 N debido a la separación eficiente de los pares de electrones y huecos, lo que restringe la recombinación de portadores fotogenerados.
En este trabajo, construimos el GaTe / C 2 N vdW heteroestructura y realiza cálculos de teoría funcional de densidad (DFT) de primeros principios para investigar sus parámetros estructurales y propiedades electrónicas y ópticas. Los resultados muestran que la heteroestructura posee una alineación de banda intrínseca de tipo II y una mejor absorción de la luz UV visible que las capas constituyentes. Además, predecimos las dependencias de deformación de la banda prohibida, las alineaciones de la banda y las posiciones del borde de la banda de GaTe / C 2 N heteroestructura, que son esenciales en el diseño de nuevos nano-dispositivos multifuncionales.
Métodos
En nuestra investigación, realizamos cálculos de primeros principios utilizando el paquete de simulación ab initio de Viena (VASP) [34]. Se adopta un conjunto de base de onda plana con un corte de energía cinética de 500 eV y un pseudopotencial de onda aumentada proyectada de Perdew-Burke-Ernzerhofer (PBE) [35] para expandir las funciones de onda y describir el potencial electrón-ión, respectivamente. Se adopta el método funcional híbrido Heyd-Scuseria-Ernzerhof (HSE06), más caro desde el punto de vista computacional [36], para corregir las bandas prohibidas subestimadas obtenidas mediante cálculos DFT / PBE. La interacción débil vdW entre las dos monocapas se describe mediante la corrección DFT-D2 de Grimme [37]. Un espacio vacío en la z Se utiliza una dirección de más de 25 Å para evitar interacciones entre heterobicapas adyacentes. A 21 × 21 × 1 (11 × 11 × 1) k -La malla para los cálculos de PBE (HSE06) se utiliza para muestrear la zona de Brillouin. Las posiciones atómicas se relajan completamente hasta que la energía y las fuerzas convergen a 10 −5 eV y 0,01 eV / Å, respectivamente.
Resultados y discusión
Comencemos por las investigaciones del prístino GaTe y C 2 N monocapas. Las configuraciones optimizadas de las dos monocapas se muestran en la Fig. 1a, b, respectivamente. Sus parámetros estructurales se enumeran en la Tabla 1. Para la monocapa de GaTe, la constante de red optimizada y la longitud del enlace Ga-Te son 4.14 y 2.41Å respectivamente. En el caso de C 2 N monocapa, la constante de celosía optimizada, las distancias C-N y C-C (1) / C-C (2) son 8.26, 1.34 y 1.47 / 1.43Å, respectivamente. Además, sus estructuras de bandas también se investigan mediante los cálculos de PBE / HSE06 y se presentan en el archivo adicional 1:Figura S1a yb, respectivamente. Aparentemente, la monocapa GaTe es un semiconductor con una banda prohibida indirecta de 1.43 / 2.13 eV mientras que C 2 La monocapa N es un semiconductor de banda prohibida directa con un valor de 1,65 / 2,44 eV. Mientras tanto, encontramos que además de un desplazamiento rígido, las estructuras de banda de C 2 La monocapa N calculada con PBE y HSE06 difieren significativamente, especialmente para las bandas de valencia. Sin embargo, los CBM y VBM calculados utilizando PBE y HSE06 están todos en Γ puntos, lo que indica que las dispersiones de banda dadas por los dos funcionales son relativamente consistentes, aunque hay alguna diferencia en la precisión. Todos los resultados concuerdan bien con los de informes anteriores [11, 38] y sugieren la fiabilidad de nuestro método de cálculo. Como es bien sabido, las bandas prohibidas de los semiconductores generalmente son subestimadas por la función PBE debido a la falta de la discontinuidad derivada en la función energética. Nuestra presentación posterior de las propiedades electrónicas y ópticas se basará en los resultados de HSE06.
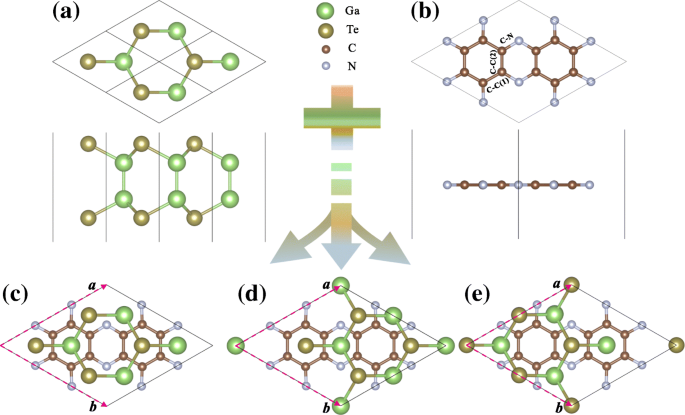
Vistas superior y lateral de ( a ) GaTe y ( b ) C 2 N monocapas. Vistas superiores de ( c - e ) α -, β - y γ -apilamiento GaTe / C 2 N heteroestructuras, en las que los vectores base correspondientes de las heteroestructuras están etiquetados
El GaTe / C 2 La heterobicapa N se construye combinando una supercélula de 2 × 2 de lámina GaTe y una celda unitaria de 1 × 1 de C 2 Capa N, con el único 0,48% de desajuste de celosía. Para encontrar la configuración estable de la heteroestructura, cambiamos la monocapa de GaTe en diferentes direcciones. Como resultado, tres tipos de apilamiento energéticamente favorables con alta simetría llamados α -, β - y γ -se obtienen apilamientos, como se ilustra en la Fig. 1c-e. En el α -apilamiento, el C 4 hexagonal N 2 los anillos están justo encima de los anillos GaTe hexagonales. En cuanto a la β - y γ -apilables, se pueden obtener moviendo la capa GaTe en el α -apila aproximadamente 1,21 y 2,42 Å a lo largo de la a + b dirección, respectivamente. Para comparar las estabilidades relativas de las tres configuraciones de apilamiento, calculamos sus energías de enlace de interfaz, \ (\ phantom {\ dot {i} \!} E _ {\ mathrm {b}} =(E _ {\ mathrm {GaTe / C_ { 2} N}} - E _ {\ text {GaTe}} - E _ {\ mathrm {C_ {2} N}}) / S \), donde \ (\ phantom {\ dot {i} \!} E _ {\ mathrm {GaTe / C_ {2} N}} \), E GaTe , y \ (E _ {\ mathrm {C_ {2} N}} \) representan las energías totales de GaTe / C 2 N heteroestructura, GaTe independiente y C 2 N monocapas, respectivamente, y S es el área de superficie de la supercélula 2D. Como se muestra en la Tabla 1, las energías de enlace de GaTe / C 2 N heteroestructuras con α -, β - y γ -Las configuraciones de apilamiento son - 15.06 meV, - 14.97 meV y - 15.80 meV / Å 2 , respectivamente. Las tres energías vinculantes están muy cerca entre sí a través de γ -El apilamiento es energéticamente más favorable, lo que es consistente con su menor distancia entre capas. Además, confirmamos las estabilidades dinámica y térmica de estas heteroestructuras con diferentes formas de apilamiento mediante el cálculo de sus espectros de fonones y la realización de simulaciones de dinámica molecular ab initio (MD) y mostramos los resultados en el archivo adicional 1:Figura S2. Todos los modos de fonón tienen frecuencias positivas excepto el modo acústico transversal cerca del Γ punto debido al ablandamiento del fonón, lo que confirma la estabilidad dinámica [5]. En las simulaciones MD, las energías totales de los sistemas oscilan en ciertos rangos de energía, y no se encuentran reconstrucciones geométricas ni enlaces rotos en las heteroestructuras, lo que indica que estos sistemas son térmicamente estables a temperatura ambiente [39]. Observamos que durante la simulación MD el γ -La configuración de apilamiento posee la menor ondulación de energía (menos de 7 meV / átomo), lo que indica su estabilidad térmica más prominente. Las energías de enlace muy cercanas de las tres configuraciones de apilamiento implican que sus estructuras electrónicas también pueden ser muy similares. Para confirmar esto, calculamos las estructuras de banda para las tres configuraciones (consulte el archivo adicional 1:Figura S3). Se puede ver que las estructuras de tres bandas son de hecho casi idénticas. Aunque el γ -La configuración de apilamiento es la más estable, las tres configuraciones aún pueden estar pobladas con algunas probabilidades a temperatura ambiente debido a sus energías de formación similares. Sin embargo, debido a que sus estructuras electrónicas también están muy próximas entre sí, solo podemos elegir una configuración para presentar nuestro trabajo. Aquí, elegimos el γ más estable -apila la configuración en los siguientes análisis y discusiones.
Pasamos ahora a las propiedades electrónicas de GaTe / C 2 Heteroestructura de N vdW. Como se muestra en la Fig. 2a, la banda prohibida de GaTe / C 2 Se calcula que N heteroestructura es de aproximadamente 1,38 eV. En comparación con el de sus componentes, su banda prohibida se reduce debido al GaTe-C 2 Interacción N y la alineación de banda resultante. Además, la estructura electrónica de C 2 La monocapa N está bien conservada. Sin embargo, la estructura de banda proyectada de GaTe en la heteroestructura tiene cambios considerables en comparación con la monocapa, lo que puede atribuirse al hecho de que la intercapa vdW y las interacciones electrostáticas pueden resultar en la superposición de estados electrónicos en las bandas de la heteroestructura. También se encuentra un comportamiento similar en MoS 2 / PbI 2 heteroestructura de vdW [40]. Además, encontramos que su VBM y CBM están localizados principalmente en GaTe y C 2 N subcapas, respectivamente. A partir de la densidad de estados total y parcial calculada (PDOS) en la Fig. 2a (panel derecho), se puede ver que el CBM se origina principalmente en p estados de los átomos de N y C, mientras que el VBM está dominado principalmente por el p estados de los átomos de Te y Ga. Las densidades de carga de banda descompuesta del CBM y VBM en la Fig. 2c, d revelan que los electrones y huecos de menor energía se distribuyen en el C 2 Capa N y capa GaTe, respectivamente, de acuerdo con los resultados detallados de PDOS anteriores. La alineación de la banda de GaTe / C 2 N heteroestructura que incluye tanto el desplazamiento VB (VBO) como el desplazamiento CB (CBO) se ilustra en la Fig. 2b, que está de acuerdo con el análisis de la Fig. 2a. Claramente, el VB y CB de la capa GaTe son más altos en energía que las bandas correspondientes del C 2 Capa N, y VBO y CBO entre GaTe y C 2 N capas son aproximadamente 1.03 y 0.72 eV, respectivamente. Cuando la heteroestructura se ilumina con luz, los electrones con energía obtenida de la luz solar saltan al CB desde el VB. Y luego, estos electrones fotogenerados en el CB de la hoja GaTe se pueden cambiar fácilmente a los del C 2 Capa de N debido a la CBO observada. Por el contrario, los agujeros fotogenerados en el VB del C 2 Transferencia de la hoja N a la de la capa GaTe debido al VBO. Los resultados anteriores sugieren que se forma una alineación de banda de tipo II en la interfaz entre GaTe y C 2 N capas, que es un requisito previo para separar los electrones y los huecos de manera eficiente. Además, la diferencia de densidad de carga promediada en el plano calculada de la heteroestructura, que se muestra en el archivo adicional 1:Figura S4, indica que algunos electrones se transfieren desde el C 2 Capa N a la capa GaTe. Significa que un campo eléctrico incorporado intrínseco ( E en ) se induce con su dirección apuntando desde C 2 Capa N a capa GaTe. También tenga en cuenta que el E en actúa en dirección opuesta (la misma) a las transferencias de electrones fotogenerados (huecos) y así inhibe la recombinación de pares electrones-huecos fotogenerados. Como resultado, bajo el efecto combinado de E intrínseco en y desplazamiento de banda, los transportes fotogenerados se pueden separar de manera efectiva en diferentes superficies, lo que puede mejorar la eficiencia de conversión de energía y, finalmente, mejorar el rendimiento de los dispositivos optoelectrónicos.
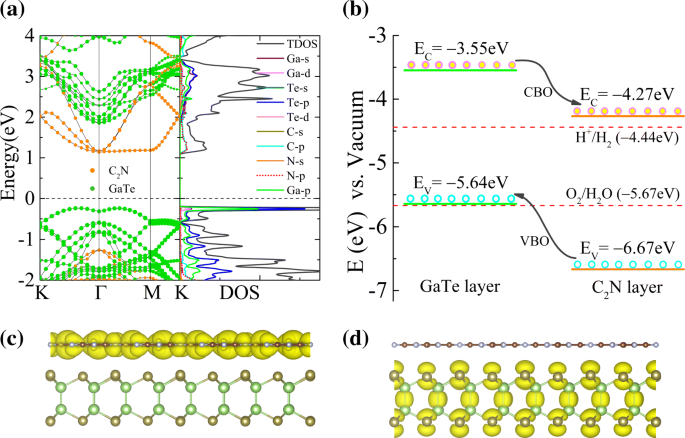
un La estructura de banda proyectada de GaTe / C 2 N heteroestructura con γ -Configuración de apilamiento y la correspondiente densidad total y parcial de estados. b Representación esquemática de alineaciones de banda de tipo II para la transferencia y separación de portadoras en GaTe / C 2 N heteroestructura, referida al nivel de vacío. Los potenciales redox (línea discontinua roja) del agua que se divide a pH =0 se muestran a modo de comparación. Banda de densidades de carga descompuesta de c VBM y d CBM de la heteroestructura
Además, notamos que el CBM de la heteroestructura se ubica más positivo que el potencial de reducción (-4,44 eV frente al nivel de vacío) del desprendimiento de hidrógeno, mientras que su VBM casi se superpone con el potencial de oxidación (-5,67 eV frente al nivel de vacío) del desprendimiento de oxígeno. Por lo tanto, solo tiene una capacidad fotocatalítica limitada para dividir el agua produciendo hidrógeno a pH =0. Sin embargo, cambiar el espaciado entre capas y el valor de pH puede encender la posible aplicación de la heteroestructura como un fotocatalizador de luz visible (ver la discusión más adelante en detalles).
En realidad, un nanodispositivo fotoeléctrico prometedor debería absorber tanta luz UV visible como sea posible. Por lo tanto, exploramos más a fondo las absorciones ópticas de GaTe / C 2 N heteroestructura y sus componentes. Los detalles computacionales se han descrito completamente en nuestros trabajos anteriores [22, 23]. Como se muestra en la Fig.3, el GaTe / C 2 La heteroestructura N exhibe una absorción de luz ultravioleta visible más fuerte y un rango de absorción más amplio en comparación con sus componentes, especialmente en el rango de energía de 2,20 a 4,71 eV. Esto se debe a las nuevas transiciones ópticas inducidas por la transferencia de carga y el acoplamiento entre capas en la heteroestructura [41].
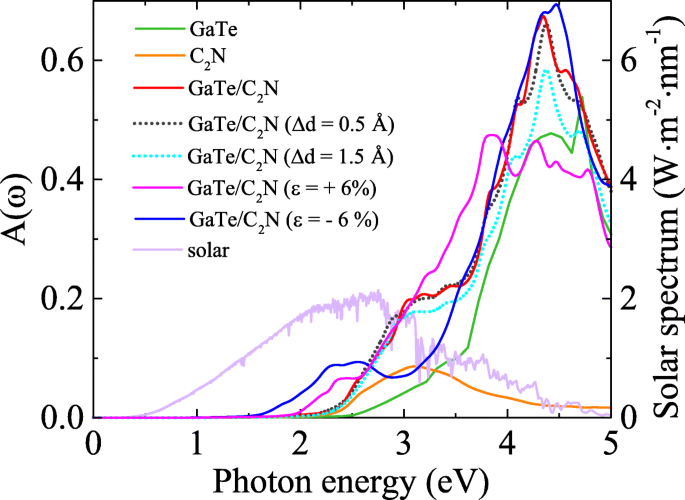
Los espectros de absorción óptica calculados A ( ω ) del GaTe / C 2 N heteroestructura y sus componentes utilizando híbrido HSE06 funcional. A ( ω ) de las heteroestructuras con deformaciones verticales de 0,5 Å y 1,5 Å y deformaciones en el plano de + 6% y -6%. Y el espectro solar también se muestra a modo de comparación
Es ampliamente conocido que las deformaciones, incluidas las deformaciones entre capas (normales) y en el plano, proporcionan una forma eficaz de ajustar las propiedades electrónicas y, por lo tanto, mejorar el rendimiento de los materiales [42]. Aquí, primero exploramos el efecto de deformación normal en GaTe / C 2 Heteroestructura de N vdW. La deformación normal se evalúa mediante Δ d = d - d 0 , donde d y d 0 son las distancias real y de equilibrio, respectivamente, entre GaTe y C 2 N subcapas. Por lo tanto, si Δ d > 0, el sistema está sometido a una deformación por tracción normal y viceversa. El cambio en la interacción entre GaTe y C 2 N capas deben reflejarse por la intensidad de la transferencia de carga entre ellas. Las diferencias de densidad de carga promediadas por plano calculadas de GaTe / C 2 N heteroestructuras con diferentes distancias entre capas se muestran en el archivo adicional 1:Figura S5. Los resultados muestran que como la distancia entre GaTe y C 2 N hojas disminuye, la transferencia de carga obviamente se intensifica como resultado de la interacción mejorada entre capas. Así, el comportamiento electrónico del GaTe / C 2 Se espera que la heteroestructura N esté bien ajustada por la tensión normal.
La banda prohibida calculada y la energía de unión de la heteroestructura como funciones de la deformación aplicada se muestran en la Fig. 4a, y las evoluciones de CBM y VBM de la heteroestructura bajo deformación normal se muestran en la Fig. 4b. Se muestra claramente que una deformación compresiva normal creciente reduce la banda prohibida debido a la interacción mejorada entre capas. Por el contrario, una deformación por tracción normal en aumento primero aumenta lentamente la banda prohibida y luego alcanza casi una convergencia en Δ d ≃0,8Å, que puede deberse a una mayor reducción de la interacción entre capas [32]. Encontramos la estructura de equilibrio en Δ d =0 tiene la energía de enlace más baja, que es consistente con el resultado que se muestra en la Tabla 1. Mientras tanto, notamos que las alineaciones de la banda de tipo II y la absorción mejorada de la luz UV visible se conservan, siendo casi independiente de la distancia entre capas (ver Fig.3 y archivo adicional 1:Figura S6). Más interesante aún, las grandes deformaciones normales de tracción ( Δ d ≃0.3 Å) desplaza el VBM debajo de O 2 / H 2 O potencial de oxidación, lo que hace que el sistema sea adecuado para la división del agua a pH =0. Durante la división del agua fotocatalítica, los procesos de producción de hidrógeno y oxígeno se producirán por separado en el C 2 Capa N y capa GaTe, respectivamente. Observamos que en tal situación, el sobrepotencial de VBM es tan pequeño que puede no ser suficiente para O 2 producción [43], pero tales potenciales de sesgo pueden ajustarse cambiando el valor de pH del medio [44]. En otras palabras, las propiedades fotocatalíticas para la división del agua se pueden modular aún más controlando el pH para que coincida con el potencial redox del agua. Como se ilustra en la Fig. 4b, en el ambiente ácido de pH =2, los bordes de la banda de la heteroestructura se encuentran perfectamente a horcajadas sobre el potencial redox del agua, mostrando que la heteroestructura es muy adecuada para H 2 / O 2 producción a partir de agua, especialmente para grandes esfuerzos verticales aplicados.
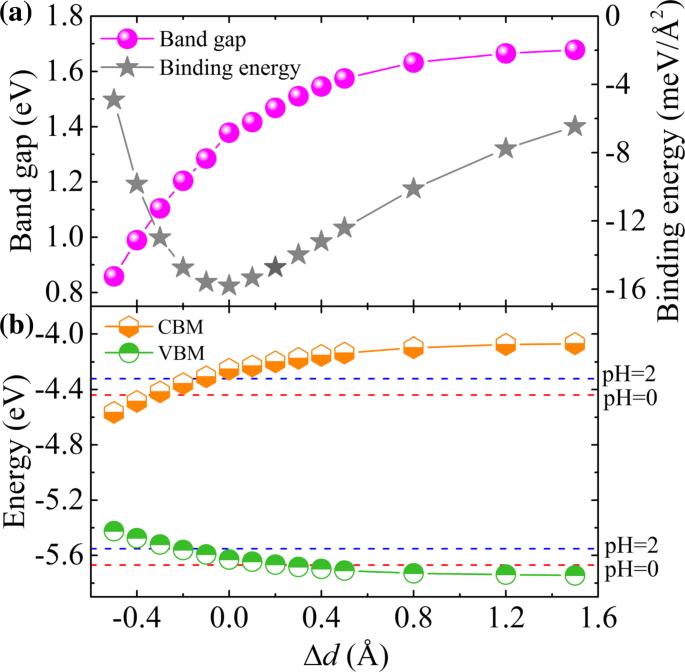
Efectos normales de tensión en a la banda prohibida y la energía de espera, y b las posiciones del borde de la banda de GaTe / C 2 Heteroestructura de N vdW. Los potenciales redox del agua que se divide a pH 0 (línea discontinua roja) y pH 2 (línea discontinua azul) se muestran a modo de comparación
Para revelar aún más el mecanismo de generación de hidrógeno fotocatalítico en GaTe / C 2 N heteroestructura, simulamos la adsorción y descomposición de agua en la superficie del C 2 Capa de N, donde se produce hidrógeno durante la división del agua fotocatalítica. Dado que la formación de moléculas de hidrógeno comienza a partir de la descomposición del agua absorbida, primero investigamos la energía de absorción de H, OH y H 2 O en el C 2 Superficie N al nivel DFT / PBE. Las energías de adsorción correspondientes son -1,03, -0,51 y -0,56 eV, respectivamente, como se ilustra en la figura 5a. Los valores negativos indican que las absorciones son energéticamente favorables [45]. Posteriormente, la energía de reacción calculada de la descomposición del agua es de aproximadamente 1,48 eV (de -0,56 a 0,92 eV). Esto significa que la descomposición del agua es una reacción endotérmica en esta superficie. Además, como los átomos de hidrógeno generados se adsorben en C 2 N de la superficie, el adatom de hidrógeno separado remotamente será energéticamente favorable para migrar cerca para formar moléculas de hidrógeno [46]. Como se muestra en la Fig. 5b, la energía de reacción requerida para eliminar un H 2 de C 2 N es relativamente pequeño (0,04 eV), lo que indica que el H 2 adsorbido es fácil de liberar y es beneficioso para la producción de gas hidrógeno fotocatalítico.
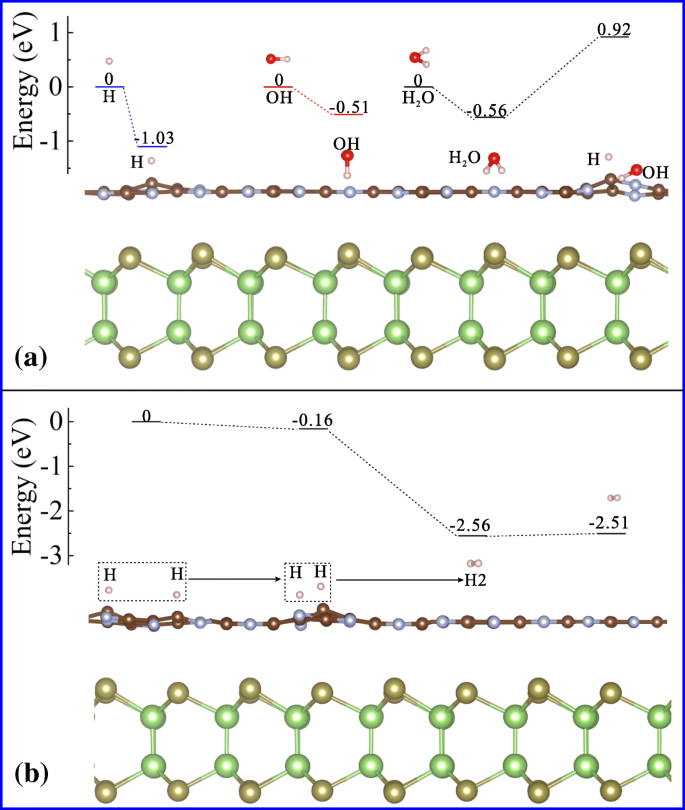
un Configuraciones de adsorción de H, OH, H 2 O y mecanismo de descomposición de H 2 O en C 2 Superficie N en GaTe / C 2 Heteroestructura de N vdW. b Interacción entre dos adatomos de hidrógeno, formación y liberación de hidrógeno molecular en C 2 Superficie N en GaTe / C 2 Heteroestructura de N vdW
Finalmente, pasamos a explorar el efecto de las deformaciones biaxiales en el plano, que se simula cambiando el parámetro de la red cristalina y se calcula mediante ε =( a - a 0 ) / a 0 , donde a y a 0 son las constantes de celosía de las estructuras deformadas y prístinas, respectivamente. Para garantizar que las deformaciones biaxiales en la capa consideradas estén dentro del rango de respuesta elástica, primero examinamos la energía de deformación por átomo, E s =( E tensado - E sin tensión ) / n , con n siendo el número de átomos en la celda unitaria. La curva de energía de deformación calculada (ver Fig. 6a (derecha y -axis)) muestra una característica de la función cuadrática, indicando que todas las deformaciones consideradas están dentro del límite elástico y, por tanto, son totalmente reversibles. La evolución de la banda prohibida bajo diversas cepas biaxiales se muestra en la Fig. 6a. Se puede ver que la banda prohibida alcanza su valor máximo (∼1,45 eV) bajo una tensión de aproximadamente -2%. En ε =- 12% el sistema experimenta una transición de semiconductor a metal, lo que implica propiedades de transporte y conducción sintonizables de esta heteroestructura. Mientras tanto, una interesante transición de banda prohibida indirecta-directa-indirecta (Ind-D-Ind) se encuentra en ε ≃− 3% y - 8%, respectivamente. Estas transiciones se derivan de los cambios de energía de banda inducidos por tensión en diferentes puntos k (consulte el archivo adicional 1 para obtener más detalles:Figura S7). La transición Ind-D y los cambios en la estructura electrónica debidos a la tensión pueden mejorar la absorción óptica [47]. En la Fig. 3, comparamos las absorciones ópticas de GaTe / C 2 N heteroestructuras bajo deformaciones de ± 6%, donde sus bandgaps son casi iguales. Los resultados muestran que las cepas biaxiales desplazan al rojo los espectros ópticos en el rango de la luz visible, lo que es coherente con la banda prohibida disminuida discutida anteriormente. Curiosamente, una deformación de - 6% conduce a una absorción óptica significativamente mejorada en la región de [1,60-2,65 eV]. Además, también se encuentra que la deformación puede cambiar la alineación de la banda. Como se muestra en la Fig. 6b y el archivo adicional 1:Figura S7, para ε ≥ + 6%, el CBM de la subcapa GaTe se desplaza hacia abajo y se convierte en el CBM de la heteroestructura. Como resultado, las energías de CBM y VBM en la subcapa GaTe están a caballo entre las de C 2 N subcapa, lo que lleva a una transición del tipo II al tipo I.Aquí, observamos que el CBM y el VBM de la subcapa GaTe se acercan entre sí bajo grandes tensiones de tracción y forman una banda prohibida muy pequeña mientras que los de C 2 La subcapa N tiene solo un cambio menor. Este comportamiento puede entenderse considerando primero los efectos de tensión en las estructuras electrónicas de las dos monocapas aisladas. Cálculos anteriores mostraron que la banda prohibida de la monocapa de GaTe es mucho más sensible a grandes deformaciones por tracción que la de C 2 N monocapa:bajo grandes tensiones de tracción, la primera se volverá muy pequeña mientras que la última permanece [11, 16]. Esto puede deberse a la estructura de pandeo de GaTe, que se ve afectada de manera más significativa por las deformaciones en el plano. Dado que las interacciones generales entre capas en la heteroestructura son débiles, principalmente el vdW y las interacciones electrostáticas que tienen solo efectos menores en la banda prohibida, los comportamientos de las dos monocapas bajo grandes tensiones de tracción se conservan en GaTe / C 2 N heteroestructura. Además, para ε ≥ − 12%, tanto el CBM como el VBM de la subcapa GaTe se vuelven más altos que los del C 2 N subcapa y, por tanto, se forma la alineación de banda de tipo III. Sin embargo, cuando la deformación por compresión aumenta aún más para ser mayor que - 13%, esta alineación de banda de tipo III se rompe, donde el C 2 La subcapa N se volverá metálica. En una palabra, la cepa puede diseñar de manera efectiva el tipo y valor de la banda prohibida y la alineación de banda de GaTe / C 2 N heteroestructura. Esto será útil para diseñar dispositivos optoelectrónicos y electrónicos multifuncionales de alto rendimiento.
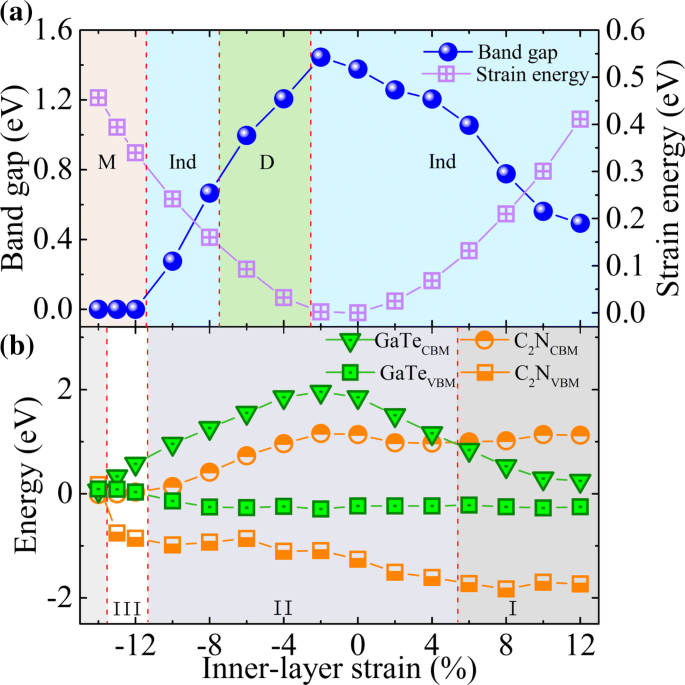
un Efectos de deformación biaxial en el plano sobre la banda prohibida y la energía de deformación de GaTe / C 2 N heteroestructura. Las regiones mistyrose, azul y verde representan los rangos de banda prohibida del metal (M), Ind y D, respectivamente. b Evolución de las posiciones del borde de la banda de las subcapas en la heteroestructura en función de la deformación biaxial en el plano. Las regiones I, II y III corresponden a alineaciones de bandas de tipo I, II y III, respectivamente
Conclusiones
En resumen, al realizar cálculos de DFT híbridos de primeros principios, hemos investigado sistemáticamente las propiedades estructurales, electrónicas y ópticas dependientes de la deformación de GaTe / C 2 N heteroestructura. Se predice que será un semiconductor de brecha indirecta que muestre absorciones ópticas mejoradas en el rango de UV visible en comparación con sus componentes. La alineación de la banda de tipo II y el campo eléctrico incorporado intrínseco inhiben la recombinación de los portadores fotogenerados con desperdicio de energía y, por lo tanto, mejoran el rendimiento de los dispositivos optoelectrónicos. En particular, las grandes tensiones de tracción normales pueden hacer que el sistema sea adecuado para la división del agua a cierto pH. Al estudiar los comportamientos de absorción y descomposición de una molécula de agua en el C 2 N subcapa en la heteroestructura, encontramos que la absorción de H 2 O y la formación de H 2 en el C 2 Las superficies de N son todas energéticamente favorables, lo que es beneficioso para producir gas hidrógeno fotocatalíticamente. Las deformaciones por compresión en el plano inducirán las transiciones Ind-D-Ind y semiconductor-metal, mientras que las deformaciones por tracción en el plano inducirán la transición de tipo II a tipo I o tipo III. Estos resultados demuestran que GaTe / C 2 La heteroestructura N tiene un gran potencial en aplicaciones de dispositivos optoelectrónicos multifuncionales.
Abreviaturas
- 2D:
-
Bidimensional
- CBM:
-
Banda de conducción mínima
- CBO:
-
Desplazamiento de la banda de conducción
- DFT:
-
Teoría funcional de la densidad
- HSE06:
-
Híbrido Heyd-Scuseria-Ernzerhof
- PBE:
-
Perdew-Burke-Ernzerhofer
- PDOS:
-
Densidad parcial de estados
- PTMC:
-
Post transition metal chalcogenides
- VBM:
-
Valence band maximum
- VBO:
-
Valence band offset
- vdW:
-
van der Waals
Nanomateriales
- Preparación y propiedades magnéticas de nanopartículas de espinela de FeMn2O4 dopadas con cobalto
- Hacia los nanofluidos de TiO2:Parte 1:Preparación y propiedades
- Estructura y propiedades electrónicas de la nanoarcilla de caolinita dopada con metal de transición
- Modulación de las propiedades de anisotropía óptica y electrónica de ML-GaS por campo eléctrico vertical
- Propiedades ópticas y electrónicas de fotodiodos N + / P de silicio hiperdopado con azufre inducido por láser de femtosegundo
- Prueba de las propiedades estructurales, electrónicas y magnéticas de Ag n V (n =1–12) Clusters
- Dependencia de la toxicidad de las nanopartículas en sus propiedades físicas y químicas
- Estructura electrónica y características I-V de las nanocintas InSe
- Propiedades de las nanopartículas de óxido de zinc y su actividad contra los microbios
- Monocapa de g-GaN adsorbido por metales alcalinos:funciones de trabajo ultrabajo y propiedades ópticas
- Propiedades de PCB automotriz y consideraciones de diseño