


Decodificación de los genomas del SARS-CoV-2 - Origen
La pandemia de COVID-19 en curso es una amenaza global para los sistemas de salud pública y ha afectado duramente a la economía mundial. El culpable de esta pandemia, el SARS-CoV-2, no es la típica gripe. Este virus afecta las vías respiratorias superiores e inferiores, interfiriendo con el proceso central de la vida, la respiración y, por lo tanto, es mortal. Hasta el 6 de abril de 2020, Worldometer ha informado 1,337,166 casos con 74,176 muertes en todo el mundo .
El examen del SARS-CoV-2 a nivel del genoma proporcionará información para comprender los orígenes de este virus. También ayudará a los científicos a diseñar herramientas de diagnóstico para detectar este patógeno invisible y facilitar la invención de terapias para minimizar la pérdida de vidas.
Comprensión del genoma del SARS-CoV-2
Un virus es un agente infeccioso que requiere un huésped vivo para prosperar y replicarse. Además, el SARS-COV-2 es un virus de ARN monocatenario con un genoma de casi 30 kb de bases de nucleótidos con 12 supuestos marcos de lectura abiertos. Poco después de que comenzara la epidemia en diciembre de 2019, los científicos chinos secuenciaron el genoma del SARS-CoV-2. Varios grupos científicos han publicado secuencias genómicas completas de SARS-CoV-2 en las últimas semanas. Estos están disponibles públicamente en Genbank y la base de datos de Coronavirus .
Origen del virus SARS-CoV-2
Durante brotes como este, las teorías de la conspiración no científicas pueden resultar en prejuicios innecesarios contra países, comunidades y culturas. El SARS-CoV-2 no es una excepción, y la situación solo se ve agravada por las crecientes plataformas de redes sociales de la actualidad. Nos incumbe ver a este enemigo invisible a través de una lente científica racional. Según los análisis del genoma, el SARS-CoV-2 es un virus que evolucionó de forma natural y no es una cepa de laboratorio sintética 1,2 . Los científicos han secuenciado los genomas completos de más de 100 cepas de SARS-CoV-2 recolectadas de diferentes regiones del mundo. Resulta que estas cepas son más del 99,5% idénticas a nivel de nucleótidos. Esto indica que las cepas no mutaron mucho en diferentes regiones, aparentemente porque el virus ya tiene una alta tasa de infección y virulencia.
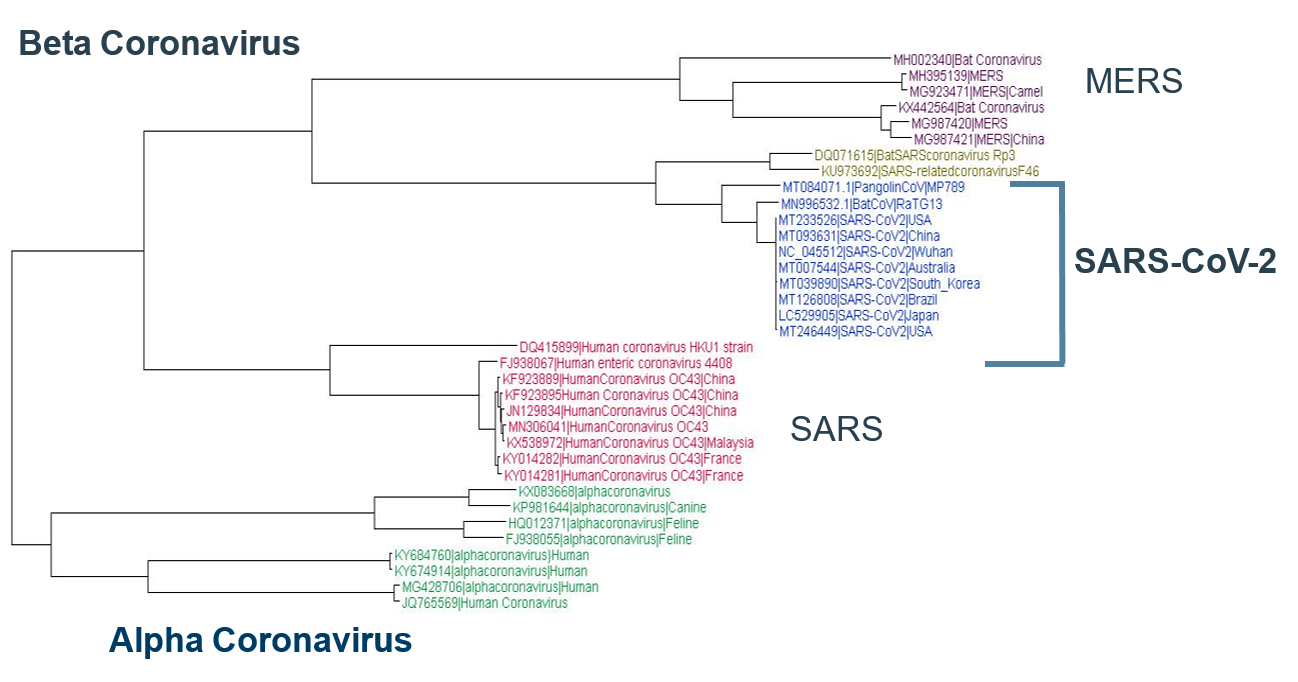
En el pasado reciente, otros dos coronavirus han recibido atención mundial. Estos fueron el SARS-CoV, China, 2002, y el MERS-CoV, Arabia Saudita, 2012. Se demostró que ambos virus anteriores se originaron en murciélagos. Basándose en este conocimiento histórico, los científicos secuenciaron el coronavirus de los murciélagos y demostraron que Bat CoV ( RaTG13 ) era 96,2% idéntico al SARS-COV-2, lo que confirma el origen zoonótico de este último. 2 El coronavirus a menudo usa un portador intermedio antes de infestar a los humanos. Curiosamente, alrededor de octubre de 2019, los informes de pangolines malayos muertos con pulmones y síntomas de fibrosis espumosa pulmonar en el centro de Rescate de Vida Silvestre de Guangdong en China llevaron a los científicos a aislar su metagenoma. De hecho, ¡los datos del metagenoma de los pangolines muertos contenían el coronavirus! 3
Curiosamente, a nivel del genoma completo, el SARS-CoV-2 es casi un 91% idéntico al malayo Pangolin CoV, lo que indica que los pangolines podrían ser un huésped intermedio.
¿Qué son los pangolines? Son mamíferos que comen hormigas y que tienen una gran demanda en Asia para su uso en la medicina tradicional china, así como para su carne, que muchos consideran un manjar. También son los mamíferos más traficados en la actualidad en el comercio ilegal de vida silvestre.
El SARS-CoV-2 es diferente de otros coronavirus conocidos, con un 88% o menos de identidad de secuencia. Según los análisis filogenéticos, el SARS-CoV-2 observado en humanos, murciélagos (RaTG13) y pangolines malayos es una nueva clase de coronavirus beta. Se han analizado casi 35 tipos diferentes de cepas de coronavirus de diferentes partes del mundo y de diferentes organismos a nivel del genoma completo. El SARS-CoV-2, que se muestra en azul a continuación, es una nueva clase de coronavirus beta (Figura 1).
¿Cómo entra el coronavirus en el anfitrión?
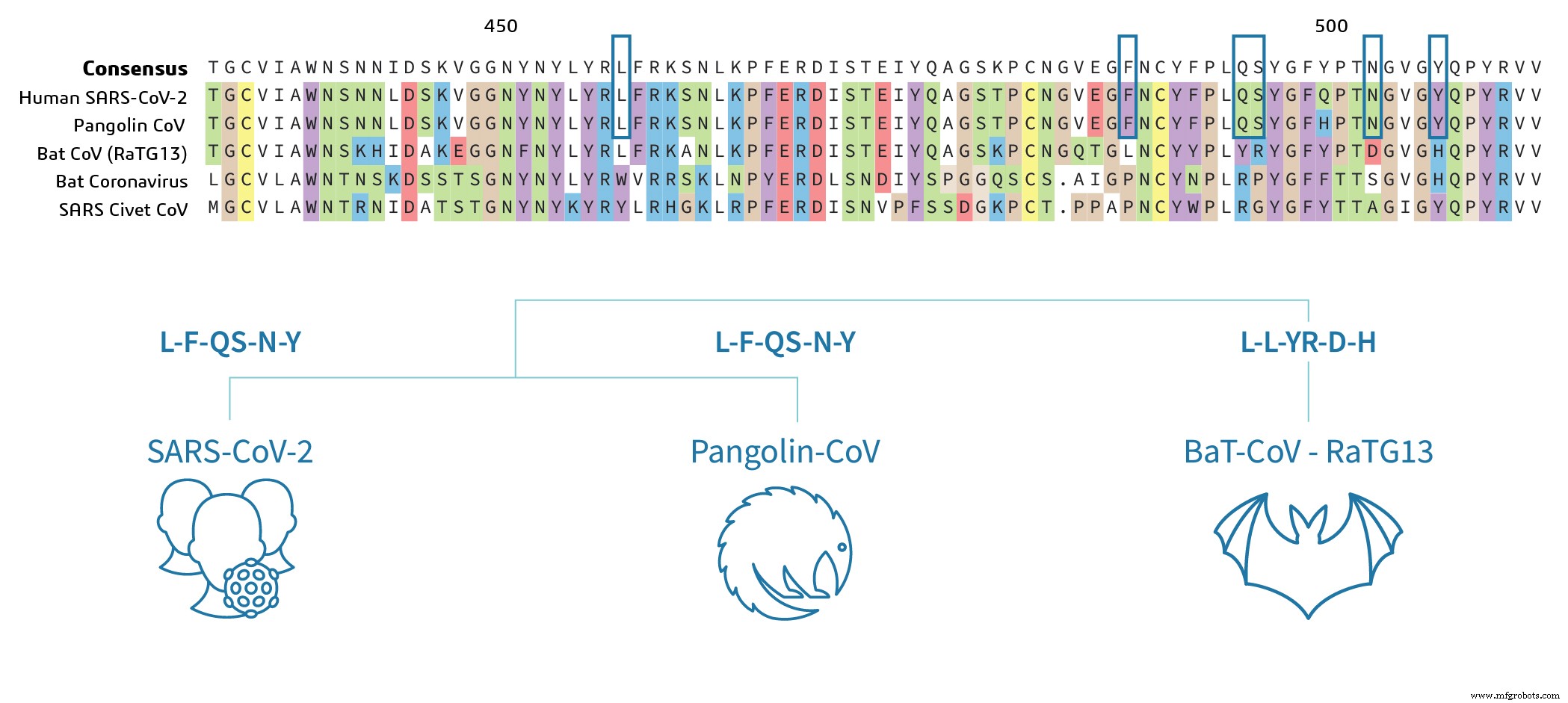
Una de las proteínas del coronavirus llamada proteína Spike parece jugar un papel importante en este proceso. La proteína Spike es una máquina molecular multifuncional que consta de dos subunidades principales, S1 y S2. La proteína Spike se une primero a un receptor en la superficie de la célula huésped a través de su subunidad S1 y luego fusiona las membranas viral y del huésped a través de su subunidad S2. El dominio en S1 de diferentes coronavirus reconoce una variedad de receptores del huésped, lo que conduce a la unión viral. El dominio de unión al receptor (RBD), que tiene 193 aminoácidos, se une y se conecta con la célula huésped. El receptor del SARS-CoV-2 en los seres humanos es la enzima convertidora de angiotensina 2 (ACE2). La ECA2 está adherida a la superficie exterior de las membranas celulares de los pulmones, las arterias, el corazón, los riñones y los intestinos. La ACE2 reduce la presión arterial al catalizar la escisión de la angiotensina II, un péptido vasoconstrictor, en angiotensina1-7, un vasodilatador. Desafortunadamente, ACE2 también parece ser un punto de entrada popular para los coronavirus.
Las secuencias de Pangolin CoV y SARS-CoV-2 están muy conservadas en la región RBD, lo que indica que el potencial patógeno del virus es muy similar entre Pangolin CoV y SARS-CoV-2. Los residuos de aminoácidos clave, que determinan la unión, son idénticos entre Pangolin CoV y SARS-CoV-2 en la alineación de secuencia (marcados con cuadros azules en la Figura 2a) y los aminoácidos clave (LFQSNY) que se muestran arriba de las caricaturas en la Figura 2b. . Curiosamente, el murciélago SARS-CoV-2 RBD difiere en 17 residuos de aminoácidos, que incluyen cinco residuos críticos para la unión de 3 . Sobre la base del análisis de los datos de la secuencia, se puede especular que el murciélago-SARS-CoV-2 puede no tener los residuos clave para unirse a la proteína ACE2 de la célula huésped para desencadenar la infección. Esto requerirá experimentación para confirmarlo.
Como se señaló anteriormente, la proteína Spike contiene dos dominios funcionales:un dominio de unión al receptor y un segundo dominio que contiene secuencias que median la fusión de las membranas viral y celular. La glicoproteína Spike debe ser escindida por proteasas celulares para permitir la exposición de las secuencias de fusión y, por lo tanto, es necesaria para la entrada celular. La comparación de la secuencia del sitio de escisión S1 / S2 de Pangolin CoV y bat-SARS-CoV-2 muestra una inserción del motivo de reconocimiento de furina. Esto indica un mecanismo distinto para la entrada del genoma viral en el citoplasma del hospedador para su replicación, como se muestra en la Figura 3.
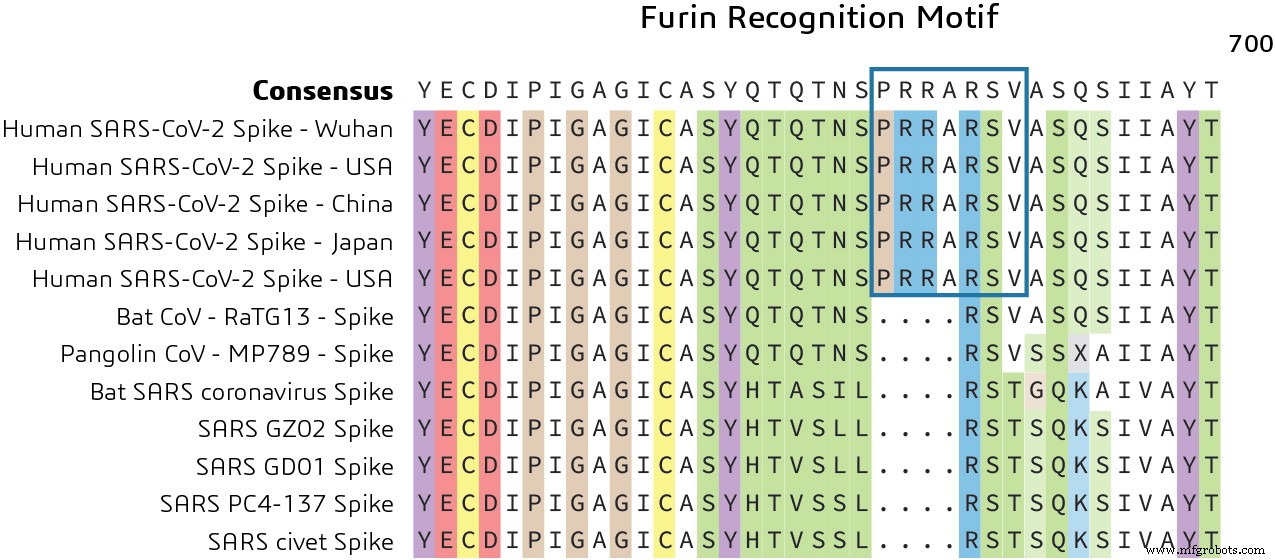
¿Cuál es el papel del motivo de reconocimiento de furin? En los seres humanos, el motivo de reconocimiento de furina (PRRARSV) es reconocido por la proteína FURIN, un miembro de la familia S8 de peptidasas de tipo subtilisina que ayuda a eliminar secciones de la proteína para cambiar su conformación de un estado inactivo a uno activo.
Se ha sugerido que la adquisición de este sitio de división de furina podría ser una "ganancia de función" que permitió a un murciélago CoV saltar a los humanos y comenzar su propagación epidémica actual. Esta podría ser una vía potencial para explorar nuevos fármacos dirigidos al bloqueo de este motivo para evitar la replicación del virus dentro del huésped.
Por lo tanto, un examen cuidadoso de la proteína Spike en el SARS-CoV-2 muestra el RBD optimizado, un motivo de reconocimiento de furina, como algunos coronavirus MERS, y su capacidad para unirse fuertemente a la proteína ACE2. Esto sugiere un proceso de selección natural en juego. Se ha demostrado que los eventos de recombinación natural en virus que co-infectan a un hospedador mejoran su rango de hospedadores, al mismo tiempo que aumentan la virulencia y la adaptación del virus. Los datos del genoma del SARS-CoV-2 con la columna vertebral del murciélago (RaTG13) y el CoV del pangolín indican nuevamente que se trata de un virus generado por recombinación natural .
¿Cuál es el donante inmediato de SARS-CoV-2 a los seres humanos?
La secuencia de SARS-CoV-2 tiene una mezcla tanto de bat-SARS-CoV (RaTG13) como de regiones de Pangolin CoV conservadas que solo pueden ocurrir durante la recombinación de estos genomas virales. Además, una ganancia de función, como se ve con el motivo de reconocimiento de furina, implica otra recombinación de virus. Para que se produzca la recombinación, es lógico que haya un huésped natural que albergue estos genomas virales. ¿Es otro pangolín? ¿U otro animal salvaje en el mercado de mariscos de Wuhan? Esto aún se desconoce. Comprender el origen podría ayudar a prevenir futuros brotes de cepas virales y pandemias globales.
Para obtener más información, contáctenos:https://www.3dsbiovia.com/about/contact/.
Biologicos
- El 555 IC
- La fórmula cuadrática
- Decodificación de "Industria 4.0"
- Un enfoque de cadena de suministro para resolver el desafío del coronavirus
- ¿La epidemia de coronavirus servirá como un llamado de atención para las cadenas de suministro globales?
- El coronavirus está destrozando las cadenas de suministro tradicionales
- Seis estrategias de cadena de suministro para petróleo y gas en la era del coronavirus
- El coronavirus podría impulsar el fin de los datos de envío incorrectos
- Cómo la ciencia de datos ayudó a combatir el brote de coronavirus
- 5 Ws del sensor SARS-CoV-2 RapidPlex
- El origen de la dobladora de tubos Crippa