


Estudio de los primeros principios sobre la estabilidad y la imagen STM del borofeno
Resumen
Muy recientemente, se ha sintetizado con éxito el borofeno (lámina de boro bidimensional atómica delgada) en la superficie de Ag (111) por deposición. Se encontraron dos tipos de estructuras. Sin embargo, la identificación de las láminas de boro monocapa que crecen sobre el sustrato metálico, así como la estabilidad de diferentes láminas de boro 2D, es controvertida. Al realizar los cálculos de los primeros principios, el presente estudio investiga la estructura atómica, la estabilidad y las propiedades electrónicas de la mayor cantidad posible de hojas de boro cultivadas en la superficie del metal, a saber, triangular pandeado, β 12 y χ 3 tipos de celosía cristalina. Nuestro resultado muestra que las tres hojas independientes son termodinámicamente inestables y todas son metálicas. Por otro lado, nuestro resultado indica que el sustrato Ag (111) estabiliza estas láminas. Además, nuestras imágenes STM simuladas de estas láminas de boro delgadas monoatómicas en la superficie de Ag (111) reproducen bien las observaciones del experimento e identifican claramente las láminas de boro recién desarrolladas.
Antecedentes
Desde el descubrimiento del grafeno, los materiales bidimensionales (2D) se han convertido en uno de los nanomateriales más activos debido a sus propiedades físicas únicas y aplicaciones potenciales en dispositivos electrónicos y de conversión de energía de próxima generación [1,2,3,4,5, 6,7]. Recientemente, se ha descubierto una clase de nanoestructuras de boro 2D que ha atraído una atención significativa [8,9,10,11,12,13,14,15,16,17,18,19,20,21]. Sin embargo, no había evidencia de que las láminas de boro 2D pudieran realizarse experimentalmente hasta hace muy poco, tanto Mannix et al. [22] y Feng et al. [23] logró avances espectaculares en la realización experimental de láminas de boro 2D delgadas atómicas. La hoja de boro 2D extendida se llama "borofeno", en analogía al grafeno.
Durante las últimas dos décadas, se han descubierto numerosas nanoestructuras de boro 2D [8,9,10,11,12,13,14,15,16,17,18,19,20,21]. Además de la hoja hexagonal y la hoja triangular [20, 21], así como las hojas triangulares dobladas [8], otras hojas de boro 2D con agujeros hexagonales, como el α -hoja [9, 18], β -hoja [9, 18], γ -hoja [19], y g1 / 8 y g2 / 15 hojas [15], fueron examinadas por los cálculos ab initio. Se sugirió que la celosía triangular plana de boro con vacantes hexagonales es más estable [9]. Y tanto los grupos de investigación computacionales como los experimentales informaron sobre una variedad de tales capas triangulares de boro con diferentes patrones de agujeros hexagonales [11, 13,14,15,16]. Sin embargo, todas estas capas de boro delgadas monoatómicas son más altas en energía que el estado general tridimensional (3D) del boro, lo que significa que la estructura 2D del boro está en desventaja termodinámicamente. Por lo tanto, es necesario un sustrato suficientemente "pegajoso" para suprimir la barrera de nucleación 3D para atraer a los átomos a la ruta 2D.
Recientemente, la formación de láminas de boro sobre metales y sustratos de boruro metálico se ha explorado mediante cálculos de primeros principios [24]; sugiere que las hojas de boro se pueden cultivar en la superficie de Ag (111) y Au (111). Además, el estudio de Piazza et al. [14] proporciona evidencia experimental de que las láminas de boro monocapa son alcanzables en base a sus observaciones de B 36 grupo; se demostró que era un cúmulo plano muy estable con un agujero hexagonal central [14]. Más recientemente, dos grupos [22, 23] sintetizaron con éxito las láminas de boro 2D cristalinas y delgadas atómicas sobre una superficie de plata evaporando directamente una fuente de boro puro mediante epitaxia de haz molecular.
Mannix y col. [22] encontró dos fases distintas de la hoja de boro sobre un sustrato de plata utilizando una caracterización de microscopía de túnel de barrido (STM) de alta resolución:una fase rayada y una fase homogénea. Feng y col. [23] también encontraron dos fases de la hoja de boro, que se ven bastante similares a las reportadas en el informe de Mannix et al., Y describieron la fase homogénea con filas en zigzag de protuberancias como χ 3 celosía de hoja de boro. Por otro lado, sus interpretaciones para la fase de franjas son bastante diferentes. Mannix y col. [22] asignó la fase rayada como una celosía triangular abrochada sin vacante. Pero Feng et al. [23] propuso que la fase de franjas sea la celosía rectangular que muestra filas paralelas de agujeros hexagonales, que se conocía como β 12 hoja.
Las configuraciones y propiedades exactas, así como las aplicaciones de estas láminas de boro 2D, han atraído una enorme atención [19, 22, 24, 25]. Se informó que el borofeno triangular abrochado es un metal altamente anisotrópico con un alto módulo de Young a lo largo de la dirección de su sillón que excede al del grafeno [22]. Sun y col. También encontró que la conductividad térmica de la red del borofeno triangular pandeado es fuertemente anisotrópica [26]. Además, Gao et al. informó que el β 12 borofeno y χ 3 El borofeno puede ser otra fase superconductora del boro además del MgB 2 película fina [27]. Sin embargo, la estabilidad termodinámica de β 12 borofeno y χ 3 borofeno son controvertidos [27, 28]. Según el estudio de Gao et al., Tanto β 12 borofeno y χ 3 el borofeno es estable [27]. Pero Penev et al. informó que tanto β 12 borofeno y χ 3 borofeno tienen frecuencias imaginarias cerca del punto G en sus espectros fonónicos [28].
Para proporcionar una mejor comprensión del borofeno alcanzable experimentalmente, investigamos sistemáticamente las posibles estructuras atómicas y su estabilidad, así como las propiedades electrónicas mediante la realización de los cálculos de los primeros principios. Nuestros resultados indican que β 12 y χ 3 las hojas son termodinámicamente inestables. Además, las configuraciones de triangular pandeado, β 12 y χ 3 todas las hojas muestran características metálicas. Además, hemos simulado las imágenes STM para la monocapa epitaxial y autónoma de boro en la superficie de Ag (111); encontramos triangular pandeado y β 12 Las hojas de boro en la superficie de Ag (111) se ven como fases de rayas pero con poca diferencia.
Métodos computacionales
Los cálculos se realizan utilizando el paquete de simulación ab-initio de Viena (VASP) basado en la teoría funcional de la densidad (DFT) [29, 30]. Se adoptó el método de onda aumentada del proyector para el cálculo de las interacciones electrón-ión [31, 32]. Y las interacciones de intercambio electrónico-correlación se describieron mediante la aproximación de gradiente generalizado (GGA) utilizando la función Perdew-Burke-Ernzerhof (PBE) [33]. Las funciones de onda se expandieron en una base de onda plana con un corte de energía de 500 eV. La primera zona de Brillion se muestreó con mallas k de 25 × 15 × 1, 15 × 9 × 1 y 11 × 11 × 1 para el triangular pandeado, β 12 y χ 3 fases de borofeno, respectivamente. Para simular las hojas de boro 2D, se incluye un espacio de vacío de al menos 20 Å a lo largo de la dirección Z para minimizar la interacción entre las imágenes periódicas. El criterio de convergencia se estableció en 10 −5 eV entre dos pasos iónicos para el proceso de autoconsistencia. Todas las estructuras se relajaron por completo hasta que la fuerza en cada átomo fue inferior a 0,02 eV Å −1 , y se fijaron las dos capas inferiores de átomos de plata. Los espectros de dispersión de fonones se han calculado utilizando el método de desplazamiento finito implementado en el paquete PHONOPY [34].
Las imágenes STM se simularon utilizando la fórmula de Tersoff-Hamann y su extensión [35]. Brevemente, asumiendo que la densidad de estados de la punta es constante, podemos aproximar la corriente de túnel STM con la densidad local de estados, \ (\ rho \ left (\ overrightarrow {r}, E \ right) \), como el única variable con la siguiente expresión:
$$ I (V) \ propto {\ int} _ {E _ {\ mathrm {F}}} ^ {E _ {\ mathrm {F}} + eV} \ rho \ left (\ overrightarrow {r}, E \ right ) dE $$ $$ \ rho \ left (\ overrightarrow {r}, E \ right) =\ sum_i \ left | {\ psi} _i {\ left (\ overrightarrow {r} \ right)} ^ 2 \ right | \ delta \ left (E- {E} _i \ right) $$donde \ (\ rho \ left (\ overrightarrow {r}, E \ right) \) es el LDOS en la superficie de la muestra, \ ({\ psi} _i \ left (\ overrightarrow {r} \ right) \) es el función de onda de muestra con energía E i y E F es la energía de Fermi. Cuando los estados en \ (\ rho \ left (\ overrightarrow {r}, E \ right) \) están llenos, también es común referirse a \ (\ rho \ left (\ overrightarrow {r}, E \ right) \) como la densidad de carga de los estados. Las imágenes STM simuladas se obtuvieron utilizando el modo de corriente constante basado en densidades de electrones calculadas.
Resultados y discusión
La Figura 1 muestra nuestros resultados calculados para el triangular pandeado, β 12 y χ 3 estructuras de celosía de borofeno. A diferencia de la configuración hexagonal plana y delgada de un átomo del grafeno, el borofeno triangular pandeado muestra pandeo a lo largo de una dirección reticular. Por otro lado, las estructuras de β 12 y χ 3 los borofenos son planos sin pandeo fuera del plano. La figura 1a muestra que hay dos átomos de boro en la celda unitaria de borofeno triangular pandeado. Y el grupo espacial de borofeno triangular abrochado es Pmmn. Nuestras constantes de celosía optimizadas son a =1.613 Å y b =2.866 Å, concordando bien con los resultados teóricos y experimentales previos [22]. El β 12 el borofeno mostrado en la Fig. 1b tiene hexágonos llenos y vacíos a lo largo de la dirección en zigzag; el grupo espacial correspondiente es P2mm. Hay cinco átomos de boro en la celda unitaria. Las constantes de celosía son 2.916 y 5.075 Å a lo largo de a y b direcciones. La celda unitaria de χ 3 el borofeno es rómbico, tiene cuatro átomos de boro y la constante de red de 4.448 Å. Su grupo espacial es C2mm. La Tabla 1 enumera los resultados de nuestros cálculos sobre las constantes de celosía, que concuerdan bien con los resultados anteriores [22, 23, 27, 36].
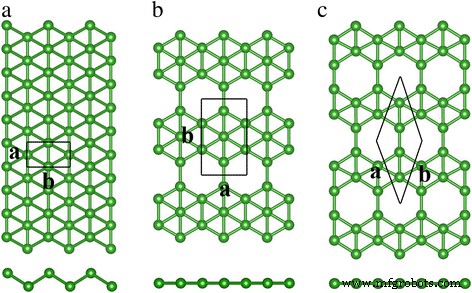
Vistas superior y lateral del triangular abrochado ( a ), β 12 ( b ) y χ 3 ( c ) hojas de boro. Las bolas verdes representan los átomos de boro. Los rectángulos y rombos encerrados por líneas negras continuas denotan las celdas unitarias. Las letras a y b representar el parámetro de celosía
Como se muestra en la Fig. 1, hay vacantes tanto en β 12 y χ 3 hojas pero no en la celosía triangular doblada y el número de vacantes en β 12 y χ 3 el borofeno es diferente. La concentración de vacantes η se define como la relación entre el número de lugares vacantes y el total de lugares (incluida la vacante) en la celda unitaria; es una cantidad que describe las láminas de boro desde puntos de vista globales y locales [9]. η es 1/6 en β 12 celosía y 1/5 de pulgada χ 3 enrejado. Comparado con el β 12 celosía, la Fig. 1c muestra que las filas de vacantes vecinas en χ 3 borofeno se desplazan a la mitad de la constante de celosía en la dirección de zigzag, lo que da como resultado un plano de simetría de C2 mm.
Calculamos la energía promedio de cada átomo de boro usando la siguiente ecuación para las tres estructuras y la usamos para comparar la estabilidad relativa de las tres estructuras; este método se ha aplicado en la Ref. [23]
$$ {E} _ {\ mathrm {FB}} ={E} _ {\ mathrm {borophene}} / n $$donde E borofeno y n son la energía y el número de átomos de boro en una celda unitaria, respectivamente. Nuestros resultados calculados se resumen en la Tabla 2. Indica que el β 12 La fase es la más estable, mientras que la χ 3 La fase es la menos estable con una energía relativamente más alta de 0.08 eV.
Luego calculamos el espectro de dispersión de fonones para las tres fases de triangular pandeado, β 12 y χ 3 borofeno. La Figura 2 muestra los espectros de dispersión de fonones a lo largo de las direcciones de alta simetría. Como se muestra en la Fig. 2a, hay tres ramas fonónicas acústicas y tres ópticas para el borofeno triangular pandeado. También muestra valores imaginarios cerca del punto G a lo largo de la dirección X – G, lo que indica que la celosía es inestable a lo largo de la a dirección, lo que explica la franja formada a lo largo de la a dirección en las imágenes STM experimentales [23]. De hecho, estudios recientes han sugerido que la tracción biaxial y la tracción uniaxial no pueden estabilizar el borofeno triangular pandeado independiente incluso bajo la tensión de tracción del 0,08% [36, 37]. La Figura 2b, c muestra que también hay frecuencias imaginarias cerca del punto G de β 12 y χ 3 etapas. Nuestros resultados muestran que las tres fases de triangular pandeado, β 12 y χ 3 son inestables.
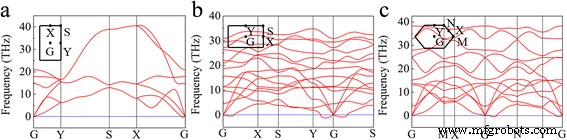
La dispersión fonética del a triangular abrochado, b β 12 y c χ 3 hojas de boro. Los puntos de alta simetría se muestran en la esquina izquierda
Estudiamos más a fondo las estructuras electrónicas del borofeno triangular pandeado, β 12 borofeno y χ 3 borofeno. Las estructuras de banda calculadas a lo largo de las direcciones de alta simetría se muestran en la Fig. 3. Como se muestra en la Fig. 3, las tres fases de triangular pandeado, β 12 y χ 3 los borofenos son metálicos. Particularmente, para el borofeno triangular pandeado como se muestra en la Fig. 3a, tres bandas de energía cruzan el nivel de Fermi:una está a lo largo de la dirección S – Y y las otras dos están a lo largo de la dirección G – X. Sin embargo, hemos mencionado en las secciones anteriores que el triangular pandeado se está pandeando a lo largo del b dirección, que abre una banda prohibida de 9,63 y 4,32 eV a lo largo de las direcciones X – S e Y – G, respectivamente. Indica que el borofeno triangular pandeado se comporta como un metal con fuerte anisotropía y la conductividad eléctrica está confinada a lo largo de la a no corrugada. dirección.
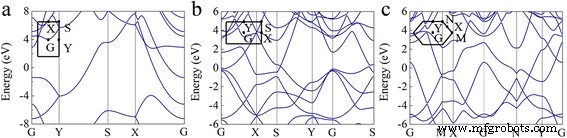
Estructuras de bandas calculadas para a triangular abrochado, b β 12 y c χ 3 hojas de boro. La energía de Fermi se puso a cero. Los puntos de alta simetría se muestran en la esquina izquierda
Además, estudiamos las estructuras atómicas y la estabilidad del triangular pandeado, β 12 y χ 3 láminas de boro sobre el sustrato Ag (111). Los resultados se muestran en la Fig. 4. La celda unitaria de borofeno triangular pandeado en la superficie de Ag (111) es la supercélula (1 × 3) de borofeno triangular pandeado independiente y la supercélula rectangular 1 × (√3) R30 ° del Ag (111) sustrato. Para la configuración de β 12 hoja en la superficie Ag (111), la celda unitaria es la celda unitaria de β 12 borofeno y 1 × (√3) supercélula R30 ° de la superficie de Ag (111). Nuestros cálculos muestran que el β 12 el borofeno coincide con la superficie de Ag (111) (~ 1% de desajuste) mejor que el borofeno triangular pandeado (~ 3% de desajuste). El χ 3 El borofeno forma dos configuraciones en la superficie de Ag (111), como se muestra en la Fig. 4c, d, que se denominan χ 3 y χ 3 ". La celda unitaria de χ 3 es un rombo con constante de celosía de a =8,67 Å, y la celda unitaria de χ 3 "Es ortorrómbica con parámetros de celosía de a =2,89 Å y b =25,02 Å; es la supercélula 1 × (5√3) R30 ° de la superficie Ag (111).
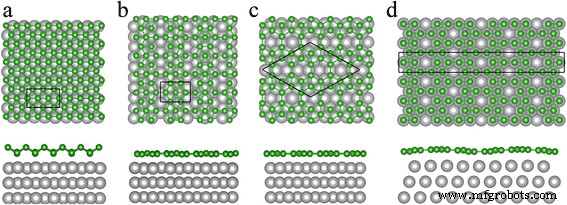
Vistas superior y lateral de láminas de boro sobre superficie Ag (111). un Triangular abrochado, b β 12 , c χ 3 y d χ 3 'Hoja de boro. Las bolas verdes y grises representan átomos de boro y plata, respectivamente. Los rectángulos y rombos encerrados por líneas negras continuas denotan las celdas unitarias de las hojas de boro en la superficie de Ag (111)
Según nuestros cálculos, las distancias verticales desde la superficie de Ag (111) a las capas atómicas de boro superior e inferior del borofeno triangular pandeado son 2,5 y 3,3 Å, respectivamente, lo que indica la débil interacción entre la hoja de boro y el sustrato de Ag. El β 12 , χ 3 y χ 3 Todas las hojas permanecen planas en la superficie de Ag (111), y las distancias verticales entre la hoja de boro y la superficie de Ag son de 2,4 ~ 2,9 Å. Los resultados concuerdan con el espesor medido de ~ 2.7 a 3.1 Å informado por Mannix et al. [22]. Comparamos las estructuras atómicas de triangular pandeado, β 12 , χ 3 y χ 3 'Fases de borofeno en sustrato Ag con las contrapartes del borofeno independiente y encontró que estas cuatro estructuras cambian poco. La altura de pandeo h de borofeno triangular pandeado es más corto de 0,910 a 0,857 Å, y las longitudes B-B son más largas alrededor de 0,1 Å. Además, las vacantes hexagonales en el β 12 el borofeno se encoge a lo largo de una dirección, y los de χ 3 borofeno se hace un poco más grande.
De manera similar al cálculo de la estabilidad relativa del borofeno independiente, calculamos además la energía promedio de cada átomo de boro para las hojas de boro en la superficie de Ag (111) mediante la siguiente fórmula:
$$ {E} _ {\ mathrm {EB}} =\ frac {1} {n} \ left ({E} _ {\ mathrm {tot}} - {E} _ {\ mathrm {sub}} \ right ) $$donde E tot es la energía total de la hoja de boro y la superficie de Ag (111), E sub es la energía del sustrato de Ag, y n es el número de átomos de boro en una celda unitaria. Nuestro resultado muestra que la posibilidad de formar triangular pandeado, β 12 , χ 3 y χ 3 Las celosías de la superficie de Ag (111) son similares en función de sus energías cercanas. Además, las energías del borofeno en la superficie de Ag (111) son menores en 0,1 ~ 0,2 eV por átomo de boro en relación con las hojas independientes. Esto significa que la superficie de Ag (111) estabiliza el borofeno.
La Figura 5 muestra nuestras imágenes STM simuladas para las hojas de boro independientes y recién desarrolladas en la superficie de Ag (111), así como la densidad de carga parcial para las hojas de boro independientes. Como se muestra en la Fig. 5a, la hoja de boro triangular abrochada independiente presenta franjas de puntos brillantes. La figura 5d indica que los puntos brillantes provienen de p z órbita de los átomos de boro superiores. La Figura 5b muestra filas de puntos redondos oscuros rodeados por hexágonos brillantes. Obviamente, las vacantes hexagonales en β 12 la celosía que se muestra en la Fig. 1b da como resultado las manchas oscuras, mientras que los hexágonos brillantes corresponden a la σ órbitas de átomos de boro alrededor de los agujeros hexagonales como se muestra en la Fig. 5e. Como se muestra en la Fig. 5c, el χ 3 la hoja muestra un patrón romboedro de puntos brillantes en forma de mancuerna. Estos puntos brillantes con mancuernas en realidad son la p z órbitas de los dos átomos de boro y el σ órbitas formadas entre ellos.
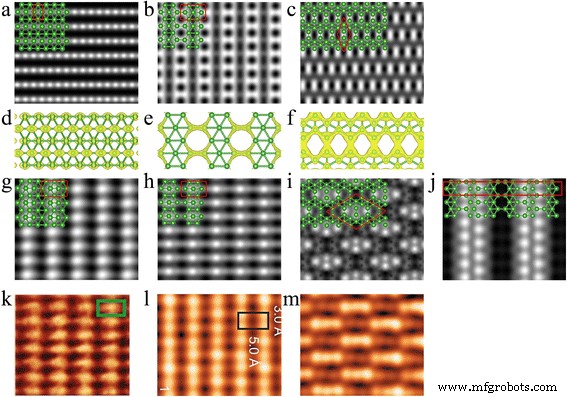
Imágenes STM simuladas de láminas de boro independientes y epitaxiales en una superficie de Ag (111). Independiente a triangular, b β 12 y c χ 3 hojas de boro. Densidad de carga parcial de d independiente triangular, e β 12 y f χ 3 hojas de boro. g Triangular abrochado, h β 12 , yo χ 3 y j χ 3 'Hoja de boro en la superficie de Ag (111). El voltaje de polarización es 1.0 V. Las bolas verdes representan los átomos de boro. Los rectángulos y rombos encerrados por líneas rojas continuas denotan las celdas unitarias de las hojas de boro independientes y recién desarrolladas en la superficie de Ag (111), respectivamente. k observado experimentalmente fase raya en Ref. [22], l fase raya en Ref. [23] y m fase homogénea en Ref. [23]
Todas las hojas de boro sobre el sustrato de Ag tienen patrones unitarios más grandes en relación con las independientes debido a los desajustes entre las celdas unitarias de borofeno y la superficie de Ag (111). La Figura 5g muestra nuestra imagen STM simulada para una hoja de boro triangular pandeada en la superficie de Ag (111). Muestra franjas de puntos brillantes en forma de huso, que concuerdan muy bien con las observaciones experimentales [22]. Comparando con la imagen de la hoja de boro triangular abrochada independiente mostrada en la Fig. 5a, la celda unitaria de la imagen STM de la hoja de boro triangular abrochada sobre la superficie de Ag (111) aumenta a tres veces. Y la forma cambia a huso de redondo. La imagen STM de β 12 La hoja sobre la superficie de Ag (111) mostrada en la Fig. 5h muestra filas de puntos ovalados oscuros rodeados por cuatro puntos brillantes en sus cuatro esquinas. Diferente de la imagen de β 12 independiente hoja que se muestra en la Fig. 5b, los puntos brillantes provienen de la p z órbitas de los átomos de boro en el centro de los hexágonos. Como se muestra en la Fig. 5i, el χ 3 la hoja tiene un patrón STM romboedro que concuerda bien con la fase S2 observada experimentalmente [23]. El grupo de puntos brillantes en la celda unitaria romboedra corresponde a la σ órbitas y p z órbitas de los átomos de boro superiores en la celda unitaria, mientras que los otros átomos de boro son invisibles porque son más bajos.
Mannix y col. [22] y Feng et al. [23] ambos informaron la fase de franjas para hojas de boro 2D en la superficie de Ag (111) basándose en sus observaciones STM, y ambas imágenes STM presentan filas paralelas de protuberancias. Sin embargo, la forma de los puntos brillantes en las dos observaciones experimentales es diferente; son fusiformes en el informe de Mannix et al. [22] y ovalados en el de Feng et al. [23]. Nuestras imágenes STM simuladas de triangular pandeado y β 12 las láminas de boro coinciden muy bien con las fases experimentales de franjas observadas en la Ref. [22] y Ref. [23], respectivamente, y las imágenes mostradas en la Fig. 5g, h reproducen claramente la diferencia entre las observaciones experimentales de Mannix et al. [22] y Feng et al. [23]. También nos proporciona una manera de distinguir las dos celosías de triangular pandeado y β 12 . En cuanto a la imagen STM de χ 3 hoja en la superficie de Ag (111), como se muestra en la Fig. 5i, concuerda con la observación experimental [23], pero nuestro resultado indica que los puntos brillantes provienen de los átomos de boro en el borde de las vacantes hexagonales en lugar de la triangular llena área como se indica en la Ref. [23].
Para distinguir aún más las estructuras de celosía de las hojas de boro en la superficie de Ag (111), simulamos las imágenes STM de la hoja de boro en Ag (111) a varios voltajes de polarización diferentes. Como se muestra en la Fig. 6, las imágenes STM simuladas para el borofeno triangular pandeado muestran franjas de puntos brillantes en forma de huso a voltaje positivo. Pero a la tensión de polarización negativa de - 0,4 V, las imágenes STM simuladas muestran las franjas claras y oscuras, lo que encaja bien con el resultado del experimento [22]. Por otro lado, las imágenes STM simuladas de β 12 el borofeno mantiene la forma ovalada tanto en el voltaje de polarización positivo como negativo. Por lo tanto, es más probable que la estructura triangular pandeada sea la configuración correcta de la fase de franjas. En cuanto a la imagen STM de χ 3 borofeno, la Fig. 6 indica que los puntos brillantes en todas las imágenes provienen de los átomos de boro en el borde de las vacantes hexagonales, pero su contraste brillante cambia a medida que el voltaje cambia de positivo a negativo. Como voltaje de polarización de 0,2 y - 0,4 V, el brillo de los puntos es similar. Además, nuestras imágenes STM simuladas para el χ 3 La configuración de 'se ve similar a la tensión de polarización de 0,8 a - 1,0 V (Fig. 6). Todos muestran los puntos brillantes que provienen de los átomos de boro en el borde de las vacantes hexagonales, pero solo los átomos de boro superiores son visibles y los átomos de boro inferiores en el medio de la celda unitaria son invisibles.

Imágenes STM simuladas para hojas de boro en Ag (111). Borofeno triangular abrochado en Ag (111) en a 0.8, e 0,2, i - 0,4 y m - 1.0 V. β 12 borofeno en Ag (111) en b 0,8, f 0,2, j - 0,4 y n - 1.0 V. χ 3 borofeno en Ag (111) en c 0,8, g 0,2, k - 0.4 y o - 1.0 V. χ 3 'Borofeno en Ag (111) en d 0,8, h 0,2, l - 0,4 y p - 1.0 V. Las bolas verdes representan los átomos de boro. Los rectángulos y rombos encerrados por líneas rojas continuas denotan las celdas unitarias de las hojas de boro recién desarrolladas en la superficie de Ag (111)
Conclusiones
En resumen, realizamos cálculos de primeros principios sobre la estructura atómica, la estabilidad y la propiedad electrónica de las tres láminas de boro 2D que se cultivaron en la superficie del metal muy recientemente, a saber, triangular pandeado, β 12 y χ 3 enrejado. Nuestros cálculos indican que las tres láminas de boro son termodinámicamente inestables sin el soporte del sustrato metálico. Las estructuras de las bandas indican que la hoja de boro triangular pandeada se comporta como un metal con fuerte anisotropía y β 12 y χ 3 Las láminas de boro también son metálicas sin huecos de energía. Además, nuestros resultados muestran que las energías para los tres tipos de celosías son muy cercanas y que la celosía coincide entre el triangular pandeado y β 12 las hojas de boro y la superficie de Ag (111) es bastante pequeña. Además, hemos encontrado que tanto triangular pandeado como β 12 las láminas de boro en el Ag (111) forman la celosía rectangular y los patrones de rayas paralelas de la imagen STM, pero con poca diferencia. Nuestros resultados proporcionan detalles para distinguir las dos celosías. Más importante aún, nuestras imágenes STM simuladas dan una nueva explicación a las hojas de boro observadas experimentalmente en la superficie de Ag (111).
Abreviaturas
- 2D:
-
Bidimensional
- 3D:
-
Tridimensional
- STM:
-
Microscopía de túnel de barrido
Nanomateriales
- Evaluación de IoT y el impacto de 5G
- Hilo de nanotubos de carbono, músculo y láminas transparentes
- Placas y hojas:¿Cuál es la diferencia?
- El estudio de un nuevo sistema micelar similar a un gusano mejorado con nanopartículas
- Estudio experimental sobre las características de flujo y transferencia de calor de nanofluidos de agua-TiO2 en un tubo estriado en espiral
- Estudio sobre la memoria de conmutación de resistencia multinivel y el fotovoltaje dependiente del estado de la memoria en uniones Pt / Nd:SrTiO3
- Diseño y ajuste de la función de trabajo del grafeno mediante tamaño, modificación, defectos y dopaje:un estudio de teoría del primer principio
- Prueba de las propiedades estructurales, electrónicas y magnéticas de Ag n V (n =1–12) Clusters
- Fabricación y caracterización de nanoclips de ZnO mediante el proceso mediado por poliol
- Nuevo estudio:impacto del COVID-19 en el futuro del trabajo y la automatización
- La diferencia entre motores de CC y CA