


Análisis por espectrometría de fluorescencia de rayos X
Análisis por espectrometría de fluorescencia de rayos X
La fluorescencia de rayos X (XRF) es una técnica espectroscópica de emisión que ha encontrado una amplia aplicación en el área de identificación y determinación de elementos. La técnica depende de la emisión de radiación X característica, normalmente en el rango de energía de 1 keV a 60 keV, después de la excitación de los niveles de energía de electrones atómicos por una fuente de energía externa, como un haz de electrones, un haz de partículas cargadas o un haz de x. -haz de rayos. En la mayoría de las matrices de muestras, la espectrometría de rayos X puede detectar elementos en concentraciones inferiores a 1 microgramo/g de muestra (1 ppm). En una muestra de película delgada, puede detectar cantidades totales de unas pocas décimas de un microgramo. Inicialmente, la espectrometría de rayos X encontró una amplia aceptación en aplicaciones relacionadas con análisis metalúrgicos y geoquímicos. Más recientemente, la espectrometría de rayos X ha demostrado ser valiosa en el análisis de muestras ambientales, en la determinación de azufre y elementos de desgaste en productos derivados del petróleo, en aplicaciones que involucran muestras forenses y en mediciones de materiales electrónicos y relacionados con la informática.
La espectrometría de fluorescencia de rayos X (XRF) es una herramienta versátil en muchos problemas analíticos. Los elementos principales, secundarios y traza se pueden determinar cualitativa y cuantitativamente en varios tipos de muestras, como metales, aleaciones, vidrios, cementos, minerales, rocas, minerales, polímeros, así como materiales ambientales y biológicos. Los elementos desde el sodio (Na) hasta el uranio (U) se determinan de forma rutinaria utilizando un espectrómetro de fluorescencia de rayos X de dispersión de energía (EDXRF), mientras que la aplicación del espectrómetro de fluorescencia de rayos X de dispersión de longitud de onda (WDXRF) permite la determinación eficiente de elementos de baja Z hasta incluso berilio (Be). Aunque las muestras se pueden analizar sin tratamiento, se pueden garantizar resultados de alta calidad si se aplica una preparación adecuada de la muestra. Esto puede variar desde una simple limpieza y pulido de la muestra (metales, aleaciones), pulverización y granulación con o sin aglutinante (cerámica, minerales, menas, suelos, etc.), fusión de la muestra con el fundente apropiado (cerámica, rocas, menas, etc.) a la digestión con ácidos (metales, aleaciones). De esta manera, los errores resultantes de la rugosidad de la superficie, el efecto del tamaño de las partículas o la falta de homogeneidad del material pueden eliminarse o minimizarse.
Roentgen descubrió los rayos X en 1895. H.G.J. Moseley desarrolló las relaciones entre la estructura atómica y la emisión de rayos X y en 1913 publicó los primeros espectros de rayos X, que son la base de la espectrometría de rayos X moderna. Moseley reconoció el potencial de las determinaciones elementales cuantitativas utilizando técnicas de rayos X. El desarrollo de la instrumentación de rayos X de rutina, que condujo al espectrómetro de rayos X conocido hoy en día, tuvo lugar durante las décadas siguientes. Coolidge diseñó un tubo de rayos X en 1913 que es similar a los que se utilizan actualmente. Soller logró la colimación de rayos X en 1924. Las mejoras en el detector de rayos X de gas por parte de Geiger y Mueller en 1928 finalmente llevaron al diseño del primer WDXRF comercial por parte de Friedman y Birks en 1948. Más recientemente, otros detectores, como el Los detectores de semiconductores de germanio y silicio dopado con litio han dado como resultado diseños de espectrómetros de rayos X modificados. La instrumentación moderna de dispersión de energía facilita la identificación cualitativa de elementos en varias muestras. El contenido de información de un espectro de rayos X de dispersión de energía se encuentra entre los más altos que se pueden obtener de materiales inorgánicos en una sola medición. La posición y la intensidad de los picos espectrales brindan información cualitativa y cuantitativa, y la intensidad del fondo brinda información sobre la composición general de la matriz de la muestra.
La espectrometría de rayos X es una de las pocas técnicas que se puede aplicar a muestras sólidas de diferentes formas. Aunque la mayoría de los espectrómetros XRF se utilizan en laboratorios, muchos están encontrando aplicación en análisis de rutina para producción y control de calidad y en tareas especializadas. En la figura 1 se muestra un diagrama estructural del espectrómetro EDXRF.
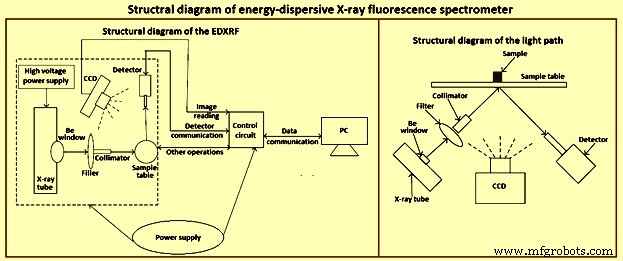
Fig. 1 Diagrama estructural del espectrómetro EDXRF
Radiación electromagnética
La radiación electromagnética es una forma de energía que puede propagarse a través del espacio y puede interactuar con átomos y moléculas para alterar su estado de energía. Ambas propiedades son importantes para la espectroscopia. La radiación electromagnética muestra un comportamiento que necesita dos teorías para explicar. La teoría ondulatoria describe el comportamiento de la radiación electromagnética, como la refracción, la reflexión, la difracción y la dispersión. La radiación se define como una forma de energía que consta de dos ondas ortogonales, cada una con la misma frecuencia y longitud de onda. Uno es un campo eléctrico oscilante y el otro un campo magnético oscilante, lo que produce el término radiación electromagnética.
En el vacío, la velocidad de propagación de la onda a través del espacio es la velocidad de la luz (c =3 × 10 elevado a 10 cm/s). Esto conduce a una importante relación fundamental representada por la ecuación w.v =c. Esta expresión establece que el producto de la longitud de onda (w) de la radiación electromagnética y su frecuencia (v) es igual a su velocidad. La longitud de onda de la radiación electromagnética varía en muchos órdenes de magnitud. Por ejemplo, las ondas de radio en la banda de transmisión AM normal tienen longitudes de onda de varios cientos de metros y las longitudes de onda ultravioleta están en el rango de 10 nm a 100 nm (nanómetro). Por el contrario, los rayos X útiles en espectroscopia oscilan entre 0,01 nm y 10 nm (Fig. 2).

Fig. 2 Rayos X y otras radiaciones electromagnéticas
Para la espectrometría de dispersión de longitud de onda, a menudo es más conveniente usar unidades de longitud de onda, pero para la espectrometría de rayos X de dispersión de energía (EDS), la descripción de la energía es más conveniente. Sin embargo, la conversión interna es simple.
Varias descripciones normalmente utilizadas de las características de los rayos X son significativas. El significado propio de la intensidad de la radiación electromagnética es la energía por unidad de área por unidad de tiempo; sin embargo, el número de conteos por unidad de tiempo del detector se usa frecuentemente como intensidad. Debido a que el área es el área activa del detector utilizado y el tiempo es un parámetro ajustable, el uso de conteos es una descripción práctica de la intensidad de los rayos X. Los términos rayos X duros o blandos se utilizan con frecuencia para diferenciar los rayos X de longitudes de onda cortas (0,01 nm a 0,1 nm) y largas (0,1 nm a 1 nm), respectivamente. La radiación X cae en la región de alta energía del espectro electromagnético.
Emisión de rayos X
Los rayos X se generan a partir de la perturbación de los orbitales electrónicos de los átomos. Esto se puede hacer de varias maneras, siendo la más común el bombardeo de un elemento objetivo con electrones de alta energía, rayos X o partículas cargadas aceleradas. Los dos primeros se utilizan con frecuencia en la espectrometría de rayos X directa o indirectamente. El bombardeo de electrones da como resultado un continuo de energías de rayos X, así como la radiación característica del elemento objetivo. Ambos tipos de radiación se encuentran en la espectrometría de rayos X.
Continuo – La emisión de rayos X con una función suave y continua de intensidad relativa a la energía se denomina radiación continua o bremsstrahlung. Un continuo de rayos X se puede generar de varias maneras. Sin embargo, el más útil es el haz de electrones que se utiliza para bombardear un objetivo en un tubo de rayos X. El continuo se genera como resultado de la desaceleración progresiva de los electrones de alta energía que inciden sobre un objetivo, que es una distribución de electrones orbitales de varias energías. A medida que los electrones que chocan interactúan con los electrones orbitales enlazados, parte de su energía cinética se convierte en radiación. La cantidad convertida depende de la energía de enlace del electrón involucrado. Por lo tanto, existe una probabilidad algo estadística de cuánta energía se convierte con cada interacción.
La probabilidad de que un electrón incidente interactúe con un electrón orbital del elemento objetivo aumenta con el número atómico del elemento, por lo tanto, la intensidad de la emisión continua aumenta con el número atómico del elemento objetivo. Además, la probabilidad de una interacción aumenta con el número de electrones por unidad de tiempo en el haz o flujo. Por lo tanto, la intensidad del continuo aumenta con la corriente del haz de electrones, expresada en miliamperios. Además, la capacidad de los electrones que chocan para interactuar con los electrones estrechamente unidos del elemento objetivo aumenta con la energía cinética de los electrones que bombardean. Dado que la energía cinética de los electrones en el haz aumenta con el potencial de aceleración, la intensidad integrada del continuo aumenta con el potencial de aceleración de electrones, expresado en kilovoltios. Finalmente, la energía máxima manifestada como fotones de rayos X es igual a la energía cinética del electrón que incide, que a su vez se relaciona con el potencial de aceleración. La energía de máxima intensidad en el continuo se encuentra alrededor de dos tercios de la energía máxima emitida. Además, existe la absorción de rayos X dentro del material objetivo o la absorción por los materiales utilizados para las ventanas del tubo de rayos X y los detectores. Por lo tanto, puede ocurrir alguna modificación de la distribución de intensidad, especialmente a bajas energías de rayos X.
Emisión característica – La mayoría de los electrones que inciden sobre un objetivo interactúan con los electrones orbitales del elemento objetivo en interacciones no específicas y producen poca o ninguna perturbación de los electrones orbitales internos. Sin embargo, algunas interacciones resultan en la eyección de electrones de estos orbitales. Las vacantes resultantes, o agujeros, representan estados inestables de alta energía. Si las vacantes orbitales están en las capas más internas, los electrones de las capas externas caen en cascada para llenarlas y esto da como resultado un estado más estable y de menor energía. La energía liberada por el proceso puede manifestarse como rayos X. Cada una de las transiciones que pueden ocurrir conducen a la emisión de líneas de rayos X nítidas características del elemento objetivo y la transición involucrada. Estas líneas de radiación características se emiten con el continuo.
absorción de rayos X
Los rayos X que inciden sobre una muestra experimentan dos interacciones importantes con los elementos de la muestra:absorción y dispersión. La absorción de la radiación puede ocurrir por interacciones específicas que son considerables en la excitación de la muestra en la espectrometría de rayos X o por interacciones más generales que influyen en la intensidad de los rayos X emitidos por la muestra. La dispersión de rayos X genera una intensidad de fondo en los espectros observados.
Absorción de masa – Cuando un haz de rayos X atraviesa un material, los fotones (campos electromagnéticos) pueden interactuar de forma no específica con los electrones en los orbitales de los elementos objetivo, reduciendo la intensidad del haz de rayos X. Las interacciones pueden provocar la eyección fotoeléctrica de electrones o la dispersión del haz de rayos X. En cualquier caso, el resultado general se describe con frecuencia en términos de una disminución exponencial de la intensidad con la longitud del camino del material absorbente. El coeficiente de absorción de masa es característico de un elemento dado a energías específicas de radiación x. Su valor varía con la longitud de onda de la radiación X y el número atómico del elemento objetivo.
El efecto fotoeléctrico es el más importante de los procesos que conducen a la absorción de rayos X cuando atraviesan la materia. El efecto fotoeléctrico es la eyección de electrones de los orbitales de los elementos en el objetivo de rayos X. Este proceso es frecuentemente el principal contribuyente a la absorción de rayos X y es el modo de excitación de los espectros de rayos X emitidos por los elementos de las muestras. Principalmente como resultado del proceso fotoeléctrico, el coeficiente de absorción de masa disminuye constantemente con el aumento de la energía de la radiación X incidente. La curva de absorción versus energía para un elemento dado tiene fuertes discontinuidades. Estos resultan de energías características en las que el proceso fotoeléctrico es especialmente eficiente.
Dispersión – Cuando los fotones de rayos X inciden en una colección de átomos, los fotones pueden interactuar con los electrones de los elementos objetivo para dar como resultado la dispersión de los fotones de rayos X, como se ilustra en la Fig. 3. La dispersión de rayos X de la muestra es la principal fuente de señal de fondo en los espectros obtenidos en la espectrometría de rayos X. La dispersión de los rayos X es causada principalmente por los electrones externos débilmente retenidos de los elementos. Si las colisiones son elásticas, la dispersión ocurre sin pérdida de energía y se conoce como dispersión de Rayleigh. Si es inelástico, el fotón de rayos X pierde energía para provocar la eyección de un electrón y la dispersión es incoherente. La trayectoria del fotón de rayos X se desvía y el fotón tiene una pérdida de energía o una longitud de onda más larga. Esta es la dispersión de Compton.
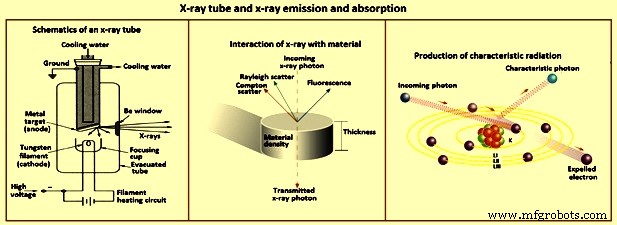
Fig. 3 Tubo de rayos X y emisión y absorción de rayos X
La dispersión afecta a la espectrometría de rayos X de dos maneras. Primero, la cantidad total de radiación dispersa aumenta con el número atómico debido a la mayor cantidad de electrones. Sin embargo, las muestras con matrices de bajo número atómico muestran una mayor dispersión observada debido a la reducción de la autoabsorción por parte de la muestra. En segundo lugar, la relación de la intensidad de dispersión 'Compton-a-Rayleigh' aumenta a medida que disminuye el número atómico de la matriz de la muestra. La pérdida de energía asociada con la dispersión Compton da como resultado un cambio predecible en la longitud de onda de la radiación.
Relaciones entre elementos y rayos X
Las diferentes relaciones entre elementos y rayos X se muestran en la Fig. 4.
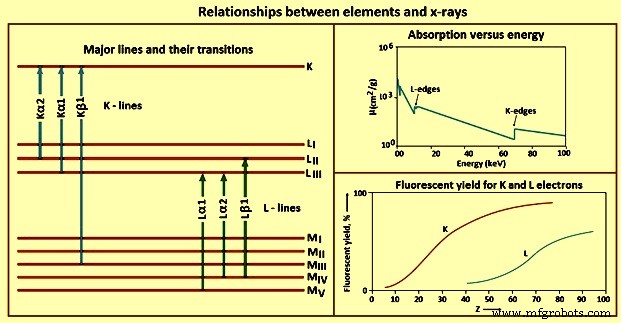
Fig. 4 Relaciones entre elementos y rayos X
Absorción – Los fotones de rayos X pueden interactuar con los electrones orbitales de los elementos para ser absorbidos o dispersados. La relación entre la absorción y el número atómico del elemento es importante para seleccionar las condiciones de funcionamiento óptimas para la espectrometría de rayos X.
Los coeficientes de absorción de masa difieren para un elemento o sustancia dada para cada elemento o sustancia a una energía dada de rayos X y a cada energía de rayos X. Debido a la mayor probabilidad de interacción con los electrones orbitales, el coeficiente de absorción de masa aumenta con el número atómico del elemento del material objetivo. A un número atómico dado, el coeficiente de absorción de masa disminuye con la longitud de onda de la radiación de rayos X. Estos resultan de energías específicas necesarias para la eyección fotoeléctrica de electrones de los diferentes orbitales del átomo y son característicos del elemento.
Los bordes de absorción son discontinuidades o puntos críticos en el gráfico de absorción de masa frente a la longitud de onda o la energía de la radiación X incidente. La energía de borde de absorción es la cantidad exacta que foto-expulsa un electrón de un orbital de un elemento. Cuanto menor sea el número cuántico principal, mayor será la energía necesaria para expulsar un electrón de esa capa. La longitud de onda de un rayo X que puede expulsar un electrón L es más larga (de menos energía) que la necesaria para expulsar un electrón de la capa K. Es decir, la energía del borde de absorción K es mayor que la energía del borde de absorción L para un elemento dado.
Emisión – El efecto fotoeléctrico es un mecanismo de absorción de rayos X mediante el cual se crean estados inestables en los orbitales electrónicos de los átomos. Una vez que se forman las vacantes en los orbitales internos, puede ocurrir la relajación al estado fundamental estable mediante la emisión de rayos X característicos del elemento excitado. La energía de los 1s El electrón está protegido del estado de los electrones de valencia, de modo que la energía del borde de absorción y la energía de los rayos X emitidos son esencialmente independientes del estado de oxidación y del enlace del átomo.
Líneas K – Una vez que el efecto fotoeléctrico crea una vacante en la capa K, el estado excitado se relaja llenando la vacante con un electrón de un orbital externo. Solo se permiten ciertas transiciones debido a las reglas de la mecánica cuántica llamadas reglas de selección. Las transiciones que siguen las reglas de selección se denominan líneas permitidas (diagrama), las que no se denominan prohibidas y las que dan como resultado átomos con dos o más vacantes en los orbitales internos en el momento de la emisión se denominan satélite (líneas no diagrama). ) líneas. El número de líneas K, y el exacto observado para un elemento, depende en parte del número de orbitales ocupados.
Líneas L – Dado que el rango práctico de energía para la mayoría de los espectrómetros de rayos X WDXRF es de 0 keV a 100 keV, y de 0 keV a 40 keV para los espectrómetros EDXRF, se debe considerar el uso de líneas de emisión distintas de las líneas K. Para un elemento dado, las líneas L se excitan con una energía de rayos X más baja que las líneas K. El uso de líneas L es particularmente valioso para elementos con números atómicos superiores a alrededor de 45.
Líneas M –Las líneas M tienen una aplicación limitada en la espectrometría de rayos X de rutina. Las líneas no se observan para elementos con números atómicos por debajo de 57, y cuando se observan, las energías de transición son bajas. El único uso práctico de estas líneas es para elementos como el torio, el protactinio y el uranio. Deben usarse solo en estos casos para evitar interferencias con las líneas L de otros elementos en la muestra.
Rendimiento fluorescente – Un electrón es expulsado de un orbital atómico por el proceso fotoeléctrico con dos posibles resultados:emisión de fotones de rayos X o eyección de electrones secundarios (Auger). Uno de estos eventos ocurre para cada átomo excitado, pero no para ambos. Por lo tanto, la producción de electrones secundarios compite con la emisión de fotones de rayos X de los átomos excitados en una muestra. La fracción de los átomos excitados que emiten rayos X se denomina rendimiento fluorescente. Este valor es una propiedad del elemento y de la línea de rayos X bajo consideración. Los elementos de número atómico bajo también tienen un bajo rendimiento fluorescente. Junto con los altos coeficientes de absorción de masa que muestran los rayos X de baja energía, la detección y determinación de elementos de bajo número atómico mediante espectrometría de rayos X es un desafío.
Efectos entre elementos – Para transiciones en espectrometría de rayos X, ninguna línea de emisión para una serie dada (K, L, M) de un elemento tiene energía igual o mayor que el borde de absorción para esa serie. Un resultado importante es que los rayos X emitidos por un elemento no pueden foto-expulsar electrones del mismo orbital de otros átomos de ese elemento. Sin embargo, una muestra compuesta por una mezcla de elementos puede mostrar interacciones que con frecuencia se denominan efectos entre elementos. Tales interacciones de elementos dentro de una muestra frecuentemente necesitan un análisis de datos especial.
Espectrómetros WDXRF
La instrumentación espectrométrica de rayos X introducida comercialmente en la década de 1950 se conoce como dispersiva de longitud de onda, lo que denota que la radiación emitida por la muestra se colima utilizando un colimador Soller y luego incide sobre un cristal de análisis. El cristal difracta la radiación en diferentes grados según la ley de Bragg y dependiendo de la longitud de onda o la energía de la radiación X. Esta dispersión angular de la radiación permite la detección secuencial o simultánea de los rayos X emitidos por los elementos de la muestra.
Los instrumentos simultáneos normalmente contienen varios conjuntos de cristales de análisis y detectores; uno se ajusta para cada analito deseado en la muestra. Aunque costosos, estos instrumentos son eficientes para la determinación rutinaria de elementos preseleccionados, pero no se convierten fácilmente para determinar elementos distintos a los seleccionados en la instalación.
Más comunes son los instrumentos secuenciales que contienen un sistema mecánico conocido como goniómetro que varía el ángulo entre la muestra, el cristal de análisis y el detector. De esta forma, la longitud de onda deseada de la radiación X puede seleccionarse mediante el movimiento del goniómetro. Los espectrómetros secuenciales WDXRF pueden controlarse por computadora para la determinación automática de muchos elementos. Las aplicaciones cuantitativas de los espectrómetros WDXRF automatizados son eficientes, ya que el instrumento se puede programar para ir a los ángulos correctos para las determinaciones deseadas. Sin embargo, las aplicaciones cualitativas son menos eficientes ya que el espectro debe explorarse lentamente.
Tubos de rayos X – Se pueden utilizar varias fuentes de energía para crear los estados electrónicos excitados en los átomos de los elementos que producen la emisión de rayos X. Entre estos se encuentran los haces de electrones, los haces de partículas cargadas y la radiación X. Los haces de electrones se dirigen sobre la muestra en técnicas tales como microscopía electrónica de barrido (SEM) y análisis de microsonda electrónica. Sin embargo, el uso de un haz de electrones necesita un alto vacío para evitar pérdidas de energía del electrón. La espectrometría de rayos X se utiliza mejor como una herramienta analítica versátil que como una herramienta especializada. Muchas muestras no son adecuadas para un alto vacío o no son conductoras, lo que provoca problemas de carga eléctrica cuando se encuentran bajo un haz de electrones. Por lo tanto, esta fuente de energía no es práctica para la espectrometría de rayos X.
Los isótopos radiactivos que emiten radiación X son otra posibilidad de excitación de los átomos para que emitan rayos X. Sin embargo, el flujo de rayos X de las fuentes isotópicas que se pueden manipular con seguridad en un laboratorio es demasiado débil para un uso práctico. Debido a que estas fuentes normalmente emiten solo unas pocas líneas estrechas de rayos X, se necesitan varias para excitar muchos elementos de manera eficiente. La fuente de energía más práctica para la espectrometría de rayos X es un tubo de rayos X (Fig. 3).
El espectrómetro WDXRF necesita una excitación eficiente de alta potencia para funcionar bien. Por lo tanto, la estabilidad y la fiabilidad del tubo de rayos X son importantes. Todos los componentes están en un alto vacío. Un filamento se calienta con un voltaje de filamento de 6 V a 14 V. El filamento calentado emite electrones térmicamente. El flujo de electrones que fluye entre el filamento y el ánodo objetivo debe ser altamente regulado y controlado. Este flujo de electrones es corriente eléctrica y normalmente se mide en miliamperios. La corriente del tubo a menudo se denomina mA.
Se aplica un potencial de varios kilovoltios entre el filamento (cátodo) y el ánodo objetivo, que sirve como potencial de aceleración para los electrones. Este voltaje se mide normalmente en kilovoltios. El ánodo es normalmente de cobre y la superficie del objetivo está recubierta con depósitos de alta pureza de elementos tales como rodio, plata, cromo, molibdeno o tungsteno. Los tubos de rayos X utilizados para la espectrometría WDXRF funcionan de 2 kW a 3 kW. Gran parte de esta energía se disipa en forma de calor, y se necesitan provisiones para enfriar el tubo de rayos X con agua. Las fuentes de alimentación y la electrónica asociada para estos tubos de rayos X son grandes. Los electrones golpean el objetivo con una energía cinética máxima equivalente al potencial del tubo aplicado. Si la energía cinética del electrón excede la energía del borde de absorción correspondiente a la eyección de un electrón orbital interno de los átomos del material objetivo, el tubo emite líneas de rayos X características del elemento objetivo. La interacción de los electrones en el haz con los electrones del elemento objetivo también conduce a la emisión de un continuo. El área del continuo y la longitud de onda de máxima intensidad dependen del potencial, la corriente y la composición del ánodo.
Análisis de cristales – Los rayos X emitidos por el tubo de rayos X se dirigen a la muestra. En la mayoría de los espectrómetros de rayos X, la muestra se coloca sobre el tubo de rayos X en lo que se conoce como óptica invertida. Esto facilita el posicionamiento de la superficie de un líquido utilizando la superficie inferior en lugar de la superior. La radiación X emitida por la muestra se colima e incide sobre la superficie de un cristal de análisis, que dispersa la radiación. El haz paralelo de radiación X policromática de la muestra se difracta desde diferentes planos de red en el cristal. El refuerzo ocurre si la distancia adicional que la radiación debe viajar por difracción desde diferentes planos de red es igual a un múltiplo entero de la longitud de onda. Si este no es el caso, se produce una interferencia destructiva. La ley de Bragg permite el cálculo del ángulo en el que se seleccionará una longitud de onda para el cristal de análisis.
Detectores – Los detectores y la electrónica asociada en el espectrómetro WDXRF detectan los rayos X difractados del cristal de análisis y rechazan las señales no deseadas, como la difracción de orden superior o inferior por el cristal de análisis o el ruido del detector. Dos detectores normalmente se colocan en tándem. El primero es un detector proporcional lleno de gas o de flujo de gas. Estos detectores consisten en un cable aislado de la carcasa. Ventanas delgadas de polímero en la parte delantera y trasera de la carcasa permiten la entrada y posible salida de la radiación X. Se aplica un potencial de polarización de unos pocos cientos de voltios entre el cable y la carcasa.
Aunque se pueden utilizar muchos gases, el gas típico es el P-10, una mezcla de 90 % de argón (Ar) y 10 % de metano. Cuando los rayos X ingresan al detector, el argón se ioniza para producir muchos pares Ar+-e-. El alambre anódico recoge los electrones y los electrones en las paredes catódicas de la carcasa neutralizan los iones Ar+. El resultado es un pulso de corriente por cada fotón de rayos X que ingresa al detector. Los detectores proporcionales llenos de P-10 son más eficientes para detectar fotones de rayos X de energías inferiores a alrededor de 8 keV (longitudes de onda superiores a alrededor de 0,15 nm). La radiación X más energética tiende a pasar a través del detector proporcional.
Un segundo detector ubicado frecuentemente detrás del contador proporcional es normalmente un detector de centelleo. Este detector consta de un cristal de yoduro de sodio dopado con talio [NaI(Tl)], que emite un estallido de luz azul (410 nm) cuando es alcanzado por un fotón de rayos X. El cristal está montado en un tubo fotomultiplicador que detecta los pulsos de luz. El número de fotones de luz producidos es proporcional a la energía del fotón de rayos X incidente. Después del procesamiento electrónico, el estallido de centelleo se convierte en un pulso de voltaje proporcional en amplitud a la energía del fotón de rayos X. Estos dos detectores se pueden operar de forma independiente o simultánea. En funcionamiento simultáneo, el potencial operativo del detector y la ganancia de salida deben ajustarse de manera que un fotón de rayos X de una energía dada produzca el mismo voltaje de altura de pulso de ambos detectores. Ambos tipos de detectores necesitan alrededor de 1 microsegundo para recuperarse entre pulsos. Algunos recuentos se pueden perder a tasas de fotones incidentes superiores a alrededor de 30.000/s. La discriminación de altura de pulso de los pulsos de rayos X del detector (es) rechaza los rayos X de orden superior o inferior difractados del cristal de análisis.
Fundamentos de funcionamiento – Cuando se considera una muestra y se selecciona el elemento analito, la primera decisión es seleccionar la línea de emisión. En ausencia de interferencias específicas, normalmente se utiliza la línea más energética plausible. Para elementos con números atómicos inferiores a 75, normalmente es la línea K, ya que muchos espectrómetros WDXRF pueden funcionar con potenciales de 100 kV para los tubos de rayos X. Cuando sea posible, se selecciona un tubo de rayos X que emita líneas características a energías justo por encima del borde de absorción para que la línea se use para el elemento analito. Cuando no se dispone de un tubo de este tipo, la excitación debe realizarse mediante el uso del continuo para un tubo de rayos X disponible.
El potencial del tubo de rayos X debe establecerse alrededor de 1,5 veces la energía del borde de absorción o más. El detector debe seleccionarse en función de la región de longitud de onda que se utilizará. El contador proporcional se utilizará para rayos X de longitud superior a 0,6 nm, el detector de centelleo para longitudes de onda inferiores a 0,2 nm y ambos para la región de superposición de 0,2 nm a 0,6 nm. Se debe seleccionar un cristal de análisis que permita detectar la longitud de onda deseada. La mayoría de las selecciones de parámetros se realizan a través del control de la computadora.
Espectrómetros de rayos X de dispersión de energía
El uso de un goniómetro en los espectrómetros de rayos X WDXRF se basa en el requisito de resolver en componentes los rayos X emitidos por varios elementos en una muestra. El uso de un dispositivo de dispersión es común en muchos tipos de espectroscopia para realizar esta tarea. Los instrumentos sin los componentes mecánicos son deseables si se puede lograr una resolución adecuada. El desarrollo de detectores de silicio derivado de litio y su aplicación a la detección de rayos X a mediados de la década de 1960 condujo a un campo de análisis espectroscópico que se conoció como espectrometría EDXRF.
Los tubos de rayos X utilizados en los espectrómetros WDXRF tienen una potencia nominal de 2 kW a 3 kW y deben enfriarse con agua. Los que se utilizan en los espectrómetros EDXRF funcionan con una potencia mucho menor y, por lo general, se enfrían con aire. Los tubos típicos varían de 9 W a 100 W. Hay diferentes materiales de ánodo disponibles y cada fabricante de espectrómetros de rayos X ofrece características especiales de tubos de rayos X. Sin embargo, después de muchas pruebas de diseño de tubos, la mayoría permanece con el diseño tradicional de "ventana lateral", aunque es mucho más pequeño que los utilizados en los espectrómetros WDXRF. Un factor importante en el diseño del tubo y la fuente de alimentación asociada es la estabilidad del tubo y el voltaje.
Una alternativa a la excitación directa del tubo de rayos X es el uso de excitación de objetivo secundario. En este modo, se usa un tubo de rayos X para irradiar un objetivo secundario, cuya característica fluorescencia de rayos X se usa a su vez para excitar la emisión de rayos X de la muestra. Debido a la pérdida sustancial de eficiencia cuando se utiliza un objetivo secundario, se necesitan tubos de rayos X de mayor potencia que los necesarios para la excitación directa.
La excitación del objetivo secundario a veces ofrece ventajas significativas. Por ejemplo, para determinar los niveles bajos de concentración de vanadio y cromo en una muestra de hierro, estos elementos se pueden excitar con un objetivo secundario de hierro sin excitar el hierro de la muestra. Con la excitación de tubo directo esto es difícil. Se necesitan varios objetivos secundarios para cubrir una amplia gama de elementos. Se ha apoyado el uso de excitación de objetivo secundario como fuente de radiación monocromática para la excitación. La importancia de esta ventaja es que muchos de los programas informáticos de parámetros fundamentales, utilizados para calcular intensidades directamente a partir de las ecuaciones básicas de rayos X, necesitan radiación de excitación monocromática.
En la práctica, la excitación del objetivo secundario solo se acerca a la radiación monocromática ideal. La excitación de tubo directo con filtros primarios apropiados funciona bien en comparación con las técnicas de objetivo secundario. Por lo tanto, la excitación directa del tubo de rayos X sigue siendo la más práctica para la mayor cantidad de aplicaciones de espectrometría de dispersión de energía (EDS). La principal fortaleza de la técnica EDS radica en sus capacidades de análisis simultáneo de elementos múltiples. Although special cases occur in which selective excitation is desirable, this frequently can be accomplished with intelligent use of an appropriate x-ray tube and filter. Any fundamental design features which limit the simultaneous multi-element capability diminish the advantage of the EDXRF spectrometer.
Since direct x-ray tube excitation is the most common method used in EDS, there are factors which govern the selection of an x-ray tube. In wavelength-dispersive techniques, several x-ray tubes are normally available for the spectrometer. These can be changed for different applications. This is not normally the case with EDS-systems, since many WDXRF spectrometer has few if any choices of primary filters. In wavelength-dispersive techniques, it is customary to attempt to excite the desired element by the characteristic emission lines of the tube anode material, but the continuum is used more efficiently in EDXRF spectrometers. The use of EDXRF spectrometers has been enhanced by computer control of tube current and voltage and selection of the primary filter. Selection and efficient use of a single x-ray tube is important in the configuration of an EDXRF system.
Characteristic lines emitted by an x-ray tube have much larger intensity at their maxima than the continuous radiation emitted. These lines are to be used for excitation whenever possible. In addition, use of a primary filter between the x-ray tube and the sample can effectively approximate monochromatic radiation impinging on the sample from these characteristic lines. EDXRF spectrometers normally offer various x-ray tube anode materials. For selecting the x-ray tube anode material, the applications most likely to be encountered are to be considered.
The principal concern is to select an anode which has characteristic lines close to, but always higher, in energy than the absorption-edge energies to be encountered. None of the characteristic lines are to create spectral interference with elements to be determined. This includes consideration of such details as the Compton scatter peak for the characteristic lines. In addition, it is difficult to perform determinations of the element of the anode material. This is especially true with samples having low concentrations of that element.
Rhodium is a favourable tube anode material for general-purpose use. The characteristic lines of this element are efficient for the excitation of elements with absorption edges to around 15 keV. The excitation efficiency for the K lines of the transition elements is low. However, the continuum can be used efficiently in this region. Rhodium also has characteristic L lines at around 2.7 keV to 3.0 keV. These are efficient for the excitation of the K lines of low atomic number elements, such as aluminum, silicon, phosphorus, and sulphur. However, in these cases, a silver anode is preferable because of the Compton scatter radiation from the rhodium lines. The characteristic lines and the continuum from the x-ray tube can be used for excitation.
Although the elements of many samples can be excited effectively using a combination of the characteristic x-ray lines from the tube anode element and the continuum, more monochromatic radiation is sometimes desired. One such situation involves enhancing the use of fundamental-parameter computations which permit quantitative determination of elements without the need for several concentration standards. A more frequent situation is the need to reduce the background in the spectrum energy range to be used in the analysis. Use of primary filters placed between the x-ray tube and the sample can be effective in these cases and are normally incorporated under computer control in commercial spectrometers.
The object is to filter the primary radiation from the x-ray tube and selectively pass the characteristic lines of the anode element. This is accomplished using a filter made of the same element as the tube anode. Since x-rays of a given line (K, L, and so on) of an element are lower in energy than the absorption edge for that element, the photoelectric component of the mass absorption coefficient is small. Such a filter does not efficiently absorb the characteristic line emitted by the x-ray tube. The higher energy x-rays from the continuum are efficient for the photoelectric process in the filter and are highly attenuated by absorption. X-rays of lower energy than the filter material absorption edge are absorbed more efficiently as the energy decreases.
The result is x-radiation striking the sample with an intensity which is largely determined by the characteristic lines of the tube anode and that approximates monochromatic radiation. Increasing the thickness of the filter decreases the total intensity, with further gain in the monochromatic approximation.
Detectors – The selective determination of elements in a mixture using x-ray spectrometry depends upon resolving into separate components the spectral lines emitted by the different elements. This process needs an energy-sorting or wavelength-dispersing device. For the WDXRF spectrometer, this is accomplished by the analyzing crystal, which needs mechanical movement to select each desired wavelength according to Bragg’s law. Optionally, several fixed-crystal channels can be used for simultaneous measurement. In contrast, EDS is based on the ability of the detector to create signals proportional to the x-ray photon energy. Hence, mechanical devices, such as analyzing crystals, are not needed.
Several types of detectors have been used, including silicon, germanium, and mercuric iodide. The solid-state, lithium-drifted silicon detector [Si(Li)] was developed and applied to x-ray detection in the 1960s. By the early 1970s, this detector was firmly established in the field of x-ray spectrometry and was applied as an x-ray detection system for SEM and x-ray spectrometry. The Si(Li) detector provides excellent resolution. It can be considered as a layered structure. Under reversed bias of around 600 V, the active region acts as an insulator with an electric-field gradient throughout its volume.
When an x-ray photon enters the active region of the detector, photo ionization occurs with an electron-hole pair created for each 3.8 eV of photon energy. Ideally, the detector is to completely collect the charge created by each photon entry and result in a response for only that energy. Some background counts appear because of energy loss in the detector. Although these are kept to a minimum by engineering, incomplete charge collection in the detector contributes to background counts. From 1 keV to 20 keV, an important region in x-ray spectrometry, silicon detectors are efficient for conversion of x-ray photon energy into charge.
Analyzer systems – The x-ray spectrum of the sample is obtained by processing the energy distribution of x-ray photons which enter the detector. One x-ray photon entering the detector causes photo-ionization and produces a charge proportional to the photon energy. Several electrical sequences are to take place before this charge can be converted to a data point in the spectrum. A detailed knowledge of the electronics is not necessary, although an understanding of their functions is important. Upon entering the Si(Li) detector, an x-ray photon is converted into an electrical charge which is coupled to a field effect transistor (FET). The FET and the electronics comprising the preamplifier produce an output proportional to the energy of the x-ray photon. Using a pulsed optical preamplifier, this output is in the form of a step signal. Since photons vary in energy and number per unit time, the output signal, due to successive photons being emitted by a multi-element sample, resembles a staircase with various step heights and time spacing. When the output reaches a determined level, the detector and the FET circuitry reset to their starting level, and the process is repeated.
The preamplifier output is coupled to a pulse processor which amplifies and shapes the signal into a form acceptable for conversion to a digital form by an analog-to-digital converter (ADC). Amplification is necessary to match the analog signal to the full-scale range of the ADC. This process involves the energy calibration of the spectrometer. Drift in the gain and/or offset (zero) of the amplification results in errors in the energy assigned to the x-ray photons producing the signal. Hence, these calibrations are to be as stable as possible, and calibration is to be routinely checked.
The energy calibration is important for qualitative identification of the elements and for precise quantitative results when using spectrum-fitting programs. The amplifier provides gain and zero controls for calibrations. Normal operation in x-ray spectrometry is to set the time on the system clock to be used to acquire the spectrum. The processing of the pulses is not instantaneous. At high count rates, the time needed can become significant. When a pulse is detected and processing initiated, the clock is ‘stopped’ until the system is ready to process a new photon. The length of time the clock is off is called dead time; the time the clock is on is called live time. Their total is real time. The system monitors live time. If the spectrometer is operated with a 50 % dead time, the real time is twice the live time.
Processing of the pulse created by a photon is to be complete before another pulse occurs. A pulse pile-up rejector circuit blocks a pulse if it is received too soon. Once activated, the pulse pile-up rejector prevents the new signal from being processed if a second x-ray enters the detector before a prior pulse is fully processed. If analysis of the prior pulse had not yet been complete, it is also to be blocked from further processing. If this blockage is not performed, pulse pile-up occurs, resulting in an artifact which appears at energies equal to the sum of the photon energy of the first and second photons to enter the detector. These are frequently called sum peaks.
Despite pulse pile-up rejection circuitry, sum peaks are observed for intense peaks in the spectrum. This is the result of two photons entering the detector simultaneously or within a time difference faster than the fast discriminator can act. Sum peaks can be observed at twice the energy of an intense peak and / or at the sum of the energies of two intense peaks in the spectrum. Sum peaks decrease rapidly in intensity with count rate. The importance of electronic pulse-processing components to system performance is easily overlooked in EDS. However, stability, linearity, and proper calibration of these components are important for the use of the spectrometer.
EDXRF spectrometers require a dedicated computer system for data acquisition. Early spectrometers were heavy, unwieldy units which used hard-wired multichannel analyzers which could acquire data, but could do little to process it. Current spectrometer and data systems based on microprocessor technology are available as table-top units.
Fundamentals of operation – The simultaneous multi-element capability of EDS complicates the selection of optimum conditions because of the factors to be considered for each element. The compromises in spectroscopy are to be made, but the initial selection of instrument operating conditions can follow a logical sequence of decisions.
Qualitative analysis needs similar procedures, normally with less stringent requirements. Once a sample is received for analysis and the elements to be determined by x-ray spectrometry are identified, the next decision is to ascertain which x-ray lines are to be used for the determinations. As a general rule, K lines are used upto a K absorption-edge energy a few keV below the characteristic line of the x-ray tube anode element. The continuum can be used for excitation if the voltage to the x-ray tube is set sufficiently high to place the continuum maximum at energy higher than the absorption edge and if a back-ground filter is used. In these cases, K absorption-edge energies can be used upto around 66 % of the maximum operating kV of the x-ray tube. However, the observed peaks lie on a continuum background and reduce the signal-to-noise ratio.
For a 50-kV x-ray tube, absorption edges as high as 30 keV can be used if the element is present in sufficient concentration. For a 30-kV rhodium or silver tube, one is restricted essentially to excitation by the characteristic tube lines. This is of no great concern unless there is a special interest in the elements between atomic numbers 41 and 50 (niobium to tin). Elements above atomic number 50 (40 for a 30-kV system) are normally to be determined using the L lines of their x-ray spectra.
To excite all L lines, the incident x-ray photon energy is to exceed the LI absorption edge. For practical use, the energy of the L lines is to be higher than around l keV. For the L line spectra, this needs atomic numbers higher than 30. At such low x-ray energies, absorption of the x-rays and low fluorescent yield in the L emission in this region needs high concentration of the element to be determined and excellent sample preparation. Overlap of the K lines of the low atomic number elements in this region also causes difficulty. For example, the K lines of phosphorus overlap with the L lines of zirconium and the M lines of iridium at around 2 keV. These problems are to be considered, but are to a large degree solved by careful use of processing software.
Once the x-ray spectral lines are selected for determination of the elements, the next step is to decide whether all analyte elements in the sample can be determined with one instrumental setting. Although the multi-element capability of EDS is useful, all elements in every sample cannot be determined with a single set of instrument parameters. Some applications need more than one condition, such as a mixture of low atomic number elements and transition elements. The transition elements are best determined by excitation using the K lines of rhodium or silver and the low atomic number elements with the L lines or a properly adjusted continuum using a background filter. Computer control of instrument parameters facilitates changing the conditions. Whether automatic or manual control is used, all samples are to be analyzed under one set of conditions, then analyzed again using the alternate set. This is preferred over changing conditions between samples.
X-ray tube operating voltage affects the efficiency of excitation of each element in the spectrum and the integrated x-ray photon flux from the tube. The tube current affects the flux only. Hence, once the operating kV has been set, the tube current typically is adjusted until the system is processing counts efficiently. System dead time is to be maintained below, but near, 50 %. The voltage and current settings for the x-ray tube have a sensitive effect on the rate of information acquisition and count distribution among the respective spectral peaks for a given type of sample.
Selection of primary tube filter thickness is important. If the filter is changed, the tube current, and sometimes the voltage, frequently needs resetting since the filter alters the intensity distribution of the x-rays striking the sample. When characteristic tube lines are used for excitation, the filter is normally made from the tube anode element. The intensity of the transmitted x-rays decrease exponentially with increasing filter thickness. It is common to have two or three primary filters made from the tube anode element in the filter holder. The selection is to reflect optimum count rate corresponding with reasonable current and voltage settings. Thicker filters attenuate lower energy radiation more effectively and reduce the excitation efficiency for the element with low absorption coefficients.
The remaining decision is the choice of atmosphere in the sample chamber. If x-rays below around 5 keV are to be implemented, use of a vacuum can be advantageous. Intensity can increase sufficiently to reduce significantly the counting time needed to achieve an adequate number of counts. If the concentration of elements yielding these x-rays is sufficiently high, the vacuum may not be needed. Because of the extra precautions needed in sample criteria and handling, a vacuum path is not to be used unless significant benefit is realized. Similar reasoning applies to the helium atmosphere.
These guidelines are useful for initial selection of operating conditions. The instrumental parameters are interactive, and a change in one parameter needs adjustment of another. For example, selection of a thicker primary filter or a decrease in the tube voltage needs an increase in the tube current.
Sample preparation
The care taken to determine the best method of sample preparation for a given material and careful adherence to that method frequently determine the quality of results obtained. Sample preparation is the single most important step in an analysis, yet it is frequently given the least attention. In most cases, the stability and overall reproducibility of x-ray instrumentation are the least significant factor affecting the precision of analytical measurements. Frequently, the precision of analytical results expected from x-ray spectrometric determinations is expressed in terms of the theoretical statistics of measurement of x-ray intensities.
When replicate samples are prepared and actual standard deviations measured, deviations are found to be larger than those predicted by counting statistics. If precision is poor, any one analytical result can also be poor, since it can differ substantially from the ‘true’ value. The variety of sample types which can be analyzed using x-ray spectrometry necessitates different sample preparation techniques.
Samples are frequently classified as infinitely thick or infinitely thin based on measurement of the attenuation of x-rays. Samples are considered to be infinitely thick if further increase in the thickness yields no increase in observed x-ray intensity. The critical value for infinite thickness depends on the energy of the emitted x-radiation and the mass absorption coefficient of the sample matrix for those x-rays. For pure iron, the critical thickness is around 40 m for iron x-rays.
Although infinitely thin samples afford several advantages, it is rarely feasible to prepare them from routine samples. Many samples fall between these two cases and need extreme care in preparation. In addition to preparation of the sample, precise positioning of the sample in the spectrometer is critical for quantitative determinations.
Solid samples – These are defined as single bulk materials, as opposed to powders, filings, or turnings. Solid samples can frequently be machined to the shape and dimensions of the sample holder. The processing is not to contaminate the sample surface to be used for analysis. In other cases, small parts and pieces are to be analyzed as received. The reproducible positioning of these samples in the spectrometer is critical. It is frequently useful to fashion a wax mould of the part which fits into the sample holder. Using the mould as a positioning aid, other identical samples can be reproducibly placed in the spectrometer.
Samples taken from unfinished bulk material frequently needs surface preparation prior to quantitative analysis. Surface finishing can be done using a polishing wheel, steel wool, or belt grinder, with subsequent polishing using increasingly fine abrasives. Surface roughness less than 100 micrometers is normally sufficient for x-ray energies above around 5 keV, but surface roughness of less than 20 micrometers to 40 micrometers is needed for energies down to around 2 keV. Several precautions are necessary. Alloys of soft metals can smear on the surface as the sample is polished, resulting in a surface coating of the soft metal which yields high x-ray intensities for that element and subsequently high analytical results.
Polishing grooves on the surface of the sample can seriously affect the measured intensity of low-energy x-rays. This can be examined by repetitive measurement of the intensity of a sample after 45 degrees or 90 degrees rotation. Use of a sample spinner reduces this effect. If a sample spinner is not available, the sample is to be placed in the spectrometer such that the incident x-radiation is parallel to the polishing direction.
Powders and briquettes – Powder samples can be received as powders or prepared from pulverized bulk material too inhomogeneous for direct analysis. Typical bulk samples pulverized before analysis are ores, and refractory materials. Powders can be analyzed using the spectrometer, pressed into pellets or briquettes, or fused with a flux, such as lithium tetra borate. The fused product can be reground and pressed or cast as a disk. For precise quantitative determinations, loose powders are rarely acceptable, especially when low-energy x-rays are used. Pressed briquettes are more reliable. However, experience indicates that the best compromise is reground and pressed fusion products. This technique eliminates several problems associated with particle-size effects.
Particle-size effects result from the absorption of the incident and emitted x-rays within an individual particle. If the mass absorption coefficient of the sample matrix is high for the x-radiation used, particles even a few microns in diameter can considerably affect attenuation of the radiation within each particle. If the sample consists of particles of various sizes, or the particle size varies between samples, the resulting x-ray intensities can be difficult to interpret. This problem is compounded by the tendency of a material composed of a mixture of particle sizes to segregate when packed.
Determination of elements using low-energy x-radiation can lead to errors from particle-size effects of as much as 50 %. If the needed speed of analysis prohibits use of fusion techniques, direct determination from packed powders can be considered. The sample is to be ground, if possible, to a particle size below the critical value. The grinding time needed frequently can be ascertained by measuring the intensity from a reference sample at increasing grinding times until no further increase is observed. The lowest energy x-ray to be used in analysis is to be selected for this test.
Briquettes or pressed powders yield better precision than packed powder samples and are relatively simple and economical to prepare. In several cases, only a hydraulic press and a suitable die are needed. In the simplest case, the die diameter is to be the same as the sample holder so that the pressed briquettes fit directly into the holder. The amount of pressure needed to press a briquette which yields maximum intensity depends on the sample matrix, the energy of the x-ray to be used, and the initial particle size of the sample. Hence, prior grinding of the sample to a particle size which is less than 100 micrometers is advisable.
A series of briquettes are to be prepared from a homogeneous powder using increasing pressure. The measured intensity of the x-ray lines to be used in the analysis is plotted versus the briquetting pressure. The measured intensity is to approach a fixed value, perhaps asymptotically. Pressures of 138 MPa to 276 MPa may be needed. For materials which do not cohere to form stable briquettes, a binding agent is needed. Acceptable binding agents include powdered cellulose, detergent powders, starch, stearic acid, boric acid, lithium carbonate, polyvinyl alcohol, and commercial binders.
Briquettes which are not mechanically stable can be improved by pressing them into backing of pre-pressed binder, such as boric acid, or by the use of a die which presses a cup from a binding agent. The sample powder can then be pressed into a briquette supported by the cup. Improved results are frequently achieved if around 0.1 mm to 0.5 mm is removed from the surface of the briquette prior to the measurement.
Fusion of materials – Fusion of materials with a flux can be performed for several reasons. Some refractory materials cannot be dissolved, ground into fine powders, or converted into a suitable homogeneous form for x-ray spectrometric analysis. Other samples can have compositions which lead to severe inter-element effects, and dilution in the flux reduces these. The fused product, cast into a glass button, provides a stable, homogeneous sample well suited for x-ray measurements. The disadvantages of fusion techniques are the time and material costs involved as well as the dilution of the elements which can result in a reduction in x-ray intensity. However, when other methods of sample preparation fail, fusion frequently provides the needed results.
Low-temperature fusions can be carried out using potassium pyro-sulphate. More common are the glass-forming fusions with lithium borate, lithium tetra-borate, or sodium tetra-borate. Flux-to-sample ratios range from 1:1 to 10:1. The lithium fluxes have lower mass absorption coefficients and hence less effect on the intensity of the low-energy x-rays. An immense variety of flux-additive recipes are reported for various sample types. Lithium carbonate can be added to render acidic samples more soluble in the flux. Lithium fluoride has the same effect on basic samples. Lithium carbonate also reduces the fusion temperature. Oxidants, such as sodium nitrate and potassium chlorate, can be added to sulphides and other mixtures to prevent loss of these elements.
Filters and ion-exchange resins – Various filters, ion-exchange resin beads, and ion-exchange resin-impregnated filter papers have become important sampling substrates for samples for x-ray spectrometric analysis. Filter materials can be composed of filter paper, membrane filters, glass fiber filters, and so on. Filters are used in a variety of applications. One widely used application is in the collection of aerosol samples from the atmosphere. Loadings of several milligrams of sample on the filter can correspond to sampling several hundred cubic meters of atmosphere. Such sampling can be performed in any environment. Many elements can be determined directly on these filters by x-ray spectrometric analysis. Particulate samples collected in this way present problems, stemming primarily from particle-size effects, which are reduced in part by the need to collect two particle-size regions using dichotomous samplers. With these units, particles are separated into those smaller and those larger than around 2 micrometers in diameter.
The smaller particles tend to represent man-made materials; the larger ones are of natural origin. The smaller particles show fewer particle-size effects, and an x-ray spectrometric determination of even low atomic number elements, such as sulphur, is possible. Glass fiber filters are frequently used for this purpose. Filters can also be used for non-aerosol atmospheric components, such as reactive gases. Filter materials can be impregnated with a reagent reactive to the gas which traps it chemically. Sampling is accomplished by conveying atmospheric gases through a treated filter under carefully controlled conditions. An example is a damp filter treated with ferric ion solution used to trap hydrogen sulphide. The excess iron can be rinsed from the filter, but the precipitated ferrous sulphide remains. The sulphur can be determined directly or indirectly by measuring the iron x-radiation. The key to determining atmospheric components is the development of suitable standards.
Filters can be used to determine solution components in ways parallel to those described for atmospheric components. Particulate materials can be filtered directly from solution. For example, particulate materials in environmental water samples are defined as that which is filtered using a 0.45 micrometer pore diameter membrane filter. Hence, filtration of particles from water can be accomplished using such filters, and direct x-ray spectrometric analysis performed. Application of filter sampling to dissolved elements in water is becoming more common. The principle is similar to the reactive reagent-impregnated filter application to atmospheric gases. In some cases, the filter can be impregnated with ion-exchange resins which trap ions as the solution passes through the filter.
Procedures using ion-exchange resin-impregnated filters are to be carefully checked, since several passes of the solution can be needed, and distribution of the ions across the paper thickness is seldom uniform. However, for solutions, a reaction can be performed prior to filtration. For example, many ions can be precipitated quantitatively from aqueous solution, even at parts per billion concentration levels. The precipitates can be collected using 0.45 micrometers pore diameter membrane filters, which are then mounted between two Mylar sheets retained by ring clips on a standard plastic sample cup. Simultaneous multi-element determinations are then performed using XRF spectrometer.
Detection limits on the filters of as low as a few tenths of a microgram are common. If 100 g of sample solution is used, this corresponds to the detection limits of a few parts per billion in the sample. Standards are easily prepared as aqueous solutions. ‘Standard reference materials’ (SRM) for environmental waters and industrial effluent water are available.
Thin-film samples – Thin-film samples are ideal for x-ray spectrometric analysis. The x-ray intensity of an infinitely thin sample is proportional to the mass of the element on the film, and the spectral intensities are free of inter-element and mass absorption coefficient effects. However, in practice, perfect thin-film samples are rarely encountered. Powder samples of sufficiently small and homogeneous particle size can be distributed on an adhesive surface, such as cellophane tape, or placed between two drum-tight layers of Mylar film mounted on a sample cup.
More important thin-film types are platings and coatings on various substrates. Analysis of these sample types is increasingly important for the electronics industry. Of particular concern are measurements of film thickness and composition. Several techniques can be used, including the substrate intensity attenuation method, the coating intensity method, various intensity ratio methods, and the variable takeoff angle method. The last method is not practical in most commercial spectrometers. To be infinitely thin to most x-rays used in x-ray spectrometric analyses, the samples are to be 10 micrometers to 200 micrometers thick.
Liquids – Liquids can also be analyzed using x-ray spectrometry. The design of x-ray spectrometric instrumentation using inverted optics, in which the sample is above the x-ray source and detector, facilitates the use of liquid samples. This convenient geometry demands caution in the preparation of liquid samples to avoid damaging the source or detector by such accidents as spills and leaking sample cups.
Quantitative standards are easily prepared for liquid samples. However, since solvents are normally composed of low atomic number elements, the Rayleigh and Compton scatter intensity is high, which increases background and leads to high limits of detection. These problems can be minimized by use of suitable primary tube filters, which reduce the scattered x-radiation in the analytically useful region.
Care is to be taken with liquids containing suspended solids. If the suspension settles during the measurement time, the x-ray intensity of the contents of the sediment is enhanced. The x-ray intensity from solution components or homogeneous suspension can decrease as a result of sediment absorption, which leads to erroneous results. This possibility is tested by brief, repetitive measurements, beginning immediately after a sample is prepared. Any observed increase or decrease in intensity with time indicates segregation in the sample. In these cases, an additive which stabilizes the suspension can be used, or the suspended content can be collected on a filter for analysis.
Special sample types – Applications of x-ray spectrometric analysis do not always provide convenient samples which can fit one of the above categories. Non-destructive analyses are occasionally needed on production products which are not 32 mm diameter circles of infinite thickness. Examples include computer disks, machined parts, and long, coated strips or wire. In these cases, a sample compartment which accommodates the samples can frequently be designed. With the development of the mercuric iodide detector, which can provide adequate resolution for many analyses without a liquid nitrogen dewar, special analytical systems for on-line and non-destructive analysis of large samples can become increasingly feasible.
Proceso de manufactura
- Análisis de forma de onda
- Opciones de análisis
- Ejemplos de circuitos y listas de red
- Análisis de fallas de componentes
- Análisis de fallas de componentes (continuación)
- ¿Qué es el análisis de red?
- Más sobre análisis de espectro
- Gafas de rayos X
- Análisis de energía impulsado por software
- Los conceptos básicos del análisis de vibraciones
- Inspección de rayos X automatizada