


Estudio de espectroscopia de absorción de rayos X sincrotrón de la estructura local en polvos BiFeO3 dopados con Al
Resumen
El BiFeO 3 dopado con Al , es decir, BFA x O muestras de polvo con x =0, 0.025, 0.05 y 0.1, se prepararon mediante la ruta hidrotermal. Los efectos de la sustitución de Al sobre las propiedades estructurales, eléctricas y ópticas de BFA x Se investigaron muestras de O. Se encuentra que la sustitución de iones Al en el sitio B de BiFeO 3 no provocó cambios estructurales y aún conserva la estructura romboédrica de perovskita con R3c simetría, que fue confirmada por las mediciones de difracción de rayos X (XRD) y Raman. Estructura fina de absorción de rayos X (XAFS) por encima de Fe K -edge y Bi L 3 -edge en BFA x También se midieron y analizaron polvos de O. Los iones Fe exhiben estados de valencia mixtos (Fe 2+ / Fe 3+ ) mientras que los iones Bi mantienen el estado de valencia + 3 en todas las muestras. Fe K -edge XAFS también indicó que había una competencia entre la hibridación de Fe 3 d y Al 3 d con O 2 p orbitales y ocurrencia de los 4 p orbitales con dopaje de Al. El Bi L 3 -edge XAFS reveló que la transición de 2 p 3/2 a 6 d estado aumentó, también lo hizo la energía de 6 d Expresar. Además, el dopaje con iones de Al afectó tanto al vecino más cercano como a las capas de coordinación del átomo de Fe y las capas vecinas más cercanas del átomo de Bi. Los resultados de la espectroscopía ultravioleta-visible (UV-Vis) muestran el BFA x El O preparado por método hidrotermal podría ser un material fotocatalítico de luz visible apropiado.
Antecedentes
Multiferroics son materiales que muestran simultáneamente propiedades ferroicas, como ferroelectricidad, ferromagnetismo y ferroelasticidad [1]. Dichos materiales muestran comportamientos interesantes como la polarización eléctrica, que se puede controlar con la aplicación de un campo eléctrico externo o viceversa. El interés en estos materiales se debe a sus amplias aplicaciones en biosensores químicos, dispositivos de almacenamiento de datos nanoelectrónicos y de alta densidad, etc. [2, 3]. Las estructuras de perovskita tienen la fórmula general ABO 3 , donde O es un ion (generalmente se refiere al oxígeno) y A y B son cationes, respectivamente. Generalmente, el catión del sitio A es más grande con un estado de valencia más bajo, que se combina con O 2- para formar una capa compacta (es decir, en la esquina de la celda unitaria). El catión del sitio B es más pequeño con un estado de valencia más alto, que puede adoptarse en un entorno de coordinación octaédrico de oxígeno (es decir, en el centro de un octaedro de aniones de oxígeno) [4]. Por definición, casi todos los multiferroicos son antiferromagnéticos (AFM) o ferromagnéticos débiles (FM) con bajas temperaturas de transición. Se dividen en dos clases:monofásicas y compuestas. Sin embargo, los materiales multiferroicos monofásicos rara vez se encuentran en la naturaleza, lo que muestra las propiedades ferroeléctricas (FE) y FM al mismo tiempo [5]. Entre todos los materiales multiferroicos conocidos hoy en día, la ferrita de bismuto (BiFeO 3 ; BFO) es uno de los materiales monofásicos que tienen un tipo de celosía de perovskita distorsionada romboédrica con un grupo espacial polar R3c . BFO muestra tanto FE con temperatura de Curie de T C ≈ 1103 ky AFM tipo G para pedidos con temperatura de Neel de T N ≈ 643 k por encima de la temperatura ambiente (RT) [6]. Este material exhibe la configuración de giro AFM tipo G a lo largo de [111] c o [001] h direcciones en su estructura pseudocúbica o romboédrica y tiene una estructura de espín en espiral superpuesta con una periodicidad de aproximadamente 62 Å a lo largo de [110] h eje en RT [7]. Tiene una gran polarización espontánea intrínseca de aproximadamente 90 μC / cm 2 atribuido a la distorsión de FeO octaédrico 6 , debido a la presencia de 6 s 2 par solitario de electrones [8]. Además, existe un acoplamiento magnetoeléctrico entre FE y parámetros de orden magnético. Las propiedades de FE dependen del par de electrones solitarios, y las propiedades de FM dependen de las capas internas parcialmente llenas, es decir, la polarización proviene del Bi-sitio (sitio A), mientras que la magnetización proviene del sitio Fe (sitio B).
Aparte de lo anterior, desafortunadamente, las principales desventajas del BFO son su baja resistividad o gran fuga de corriente, debido a los defectos de carga tales como vacantes de bismuto y oxígeno, fases de impurezas, fluctuaciones de valencia del hierro y mala calidad interfacial [9]. Además, es difícil obtener BFO de alta calidad debido a algunas fases de impurezas como Bi 2 Fe 4 O 9 (grupo espacial Pbam ) y Bi 25 FeO 39 (grupo espacial I23 ). Es inevitable que las impurezas se generen en el proceso de preparación. Para abordar estos problemas y limitaciones, varios grupos de investigación han estado utilizando varios métodos para superar los defectos del BFO, por ejemplo, modificación de la cepa, sustitución del dopaje con iones divalentes y de tierras raras. Ahora, dentro de este campo de estudio, con el dopaje de iones de metales de transición o elementos de tierras raras en el sitio A o en el sitio B, o el dopaje conjunto en el sitio A y B, se pueden mejorar las propiedades multiferroicas del BFO. Por ejemplo, el dopaje con elementos de tierras raras puede estabilizar la estructura de la perovskita, retener la no centrosimetría y controlar la vaporización de Bi 3+ iones [10]. El dopaje con iones de metal de transición puede reducir la fluctuación de valencia del Fe 3+ iones. Ya se han informado elementos como Pr, Sm, Eu, Gd y La [11, 12] para la sustitución del sitio A y Mn, Cr y Ti [13,14,15] para la sustitución del sitio B. Además, las propiedades magnéticas, dieléctricas y ferroeléctricas se pueden mejorar con el codopaje. Para el codopaje de sitios A y B de BFO, se han informado La-Gd, Ba-Ni, Dy-Cr, Y-Mn y Tb-Ti [16,17,18,19,20]. Hasta ahora, varias rutas que incluyen sol-gel [21], mecanoquímico [22], autocombustión [23], deposición por láser pulsado [24] , e hidrotermales [25, 26] se han informado para preparar BFO. El método hidrotermal se ha aplicado ampliamente debido a sus propiedades de ahorro de energía, dispersión fina, bajo costo y tamaño de partícula pequeño [27]. La temperatura de reacción debe ser lo suficientemente alta para formar BFO y también debe usarse para eliminar las fases secundarias durante la preparación de la muestra. La mayoría de los estudios previos de Al dopado tanto en el sitio A como en el sitio B del BFO han sido investigados por las propiedades estructurales, ópticas y de transporte de Azam et al [28]. Madhu y col. [29] han informado de las aplicaciones fotocatalíticas del BFO dopado con Al en el sitio B. Otro informe de Jawad et al. [30] exploró el comportamiento dieléctrico de las cerámicas BFO nanoestructuradas. El trabajo de Wang et al. [31] estudió en detalle los cristales huecos de BFO dopado con Al. Sin embargo, aún no se conocen algunas propiedades físicas importantes, como los efectos del dopaje en el sitio B sobre la estructura electrónica local de los materiales. En la etapa actual, la espectroscopia de estructura fina de absorción de rayos X (XAFS) es una de las formas poderosas de estudiar el entorno local de un átomo y proporciona información estructural de los materiales, así como energía de absorción, estado de valencia del elemento, transferencia de carga, etc. y tipo de unión [32]. Hasta donde sabemos, no se han encontrado informes sobre el efecto del dopaje de Al en el sitio B sobre la estructura electrónica local de la investigación de BFO por parte de XAFS.
En este trabajo, el BFO sin dopar y las composiciones objetivo BiFe 1- x Al x O 3 (BFA x O) con x =0, 0.025, 0.05 y 0.1 se sintetizaron por vía hidrotermal. El foco principal es la investigación del efecto del dopaje con Al en el sitio B sobre las propiedades del BFO que se comparan con el BFO sin dopar. Las propiedades estructurales se investigaron en detalle.
Métodos
Se utilizó el método hidrotermal para obtener BFO y BFA sin dopar x O muestras. Los reactivos químicos utilizados en este trabajo fueron nitrato de bismuto (Bi (NO 3 ) 3 · 5H 2 O), nitrato de hierro (Fe (NO 3 ) 3 · 9H 2 O), nitrato de aluminio (Al (NO 3 ) 3 · 6H 2 O) e hidróxido de potasio (KOH). Todos los reactivos químicos se usaron tal como se recibieron sin purificación adicional. Bi (NO 3 ) 3 · 5H 2 O y Fe (NO 3 ) 3 · 9H 2 O se utilizaron como materiales de origen, mientras que Al (NO 3 ) 3 · 6H 2 Se utilizaron O y KOH como aditivos. Se utilizó agua desionizada para preparar todas las soluciones acuosas. Una corrida típica para preparar polvos de BFO es la siguiente:Los 20 ml de Bi (NO 3 ) 3 · 5H 2 O, Fe (NO 3 ) 3 · 9H 2 O y Al (NO 3 ) 3 · 6H 2 O se colocaron en un autoclave de acero inoxidable de 80 ml y se mezclaron bien. Después de eso, se goteó lentamente una cantidad apropiada de solución de KOH a la solución de mezcla anterior hasta que se llenó el 65-80% de su volumen, que luego se transfirió al aparato de agitación magnética fuerte para agitar durante 2 ~ 3 ha 80 ° C para Consiga una solución clara. De acuerdo con el procedimiento del método, la solución de color marrón oscuro obtenida se transfirió a un autoclave de acero inoxidable revestido con teflón. El tratamiento hidrotermal se llevó a cabo a una temperatura de 200 ° C durante 10 h bajo presión autógena. La velocidad de calentamiento fue de 2 ° C / min. Una vez completada la reacción hidrotermal, los productos resultantes se enfriaron a TA de forma natural. Posteriormente, los polvos resultantes se recolectaron y lavaron varias veces con acetona, agua desionizada y etanol hasta que el valor de pH de las soluciones alcanzó 7. Finalmente, el BFA x Los polvos O se colocaron en un horno de secado con termostato durante 6 ha 70 ° C y luego se secaron para su caracterización adicional. Hemos preparado cuatro conjuntos de muestras de BFA x O variando la concentración de Al (NO 3 ) 3 · 6H 2 O de 0–0,1 M.
La estructura cristalina de BFA x Las muestras de O se determinaron mediante difracción de rayos X (XRD, Mac Science M18XHF22-SRA). Se empleó espectroscopía Raman (Renishaw InVia Reflex) con radiación de un láser Ar + para determinar las propiedades estructurales de los polvos a temperatura ambiente. Los datos XAFS se recopilaron en el modo de transmisión a varias concentraciones en la línea de luz 1W2B de la Instalación de Radiación Sincrotrón de Beijing (BSRF), China. Fe K -espectro de borde con una resolución energética de Δ E / E :2 × 10 −4 y Bi L 3 -espectro de borde con una resolución energética de Δ E / E :1 × 10 −4 se midieron para el BFA x O muestras a TA. Para obtener los mejores datos XAFS, BFA x Los polvos O se molieron en un mortero de ágata, luego se mezclaron con BN y finalmente se comprimieron en gránulos. La corrección de fondo, la normalización y la región anterior al borde y posterior al borde del espectro de absorción fueron ajustadas por ATHENA, un software para el procesamiento de datos XAFS, dentro del programa IFEFFIT [33]. El E o El valor fue determinado por el máximo en la primera derivada en la región del borde. Extrajimos el χ ( k ) perfil en el k espacio de 0–12 Å −1 . El k 3 × χ ( k ) perfil se transformó Fourier a la R espacio de 0–8 Å, utilizando la función de ventana de Hanning. El Fe 2 O 3 y Bi 2 O 3 se midieron como compuestos de referencia. Las propiedades ópticas de los polvos se evaluaron usando espectrofotómetro ultravioleta-visible (UV-Vis, UV 3900H). En este trabajo, la investigación se limita a la baja concentración de dopaje de 0 ≤ x ≤ 0,1.
Resultados y discusión
Los patrones XRD del BFO sin dopar ( x =0) y BFA x O polvos, que escanearon de 2 θ valor de 15-60 °, se muestran en la Fig. 1. Los espectros completos de los patrones de XRD en la Fig. 1a indican que todas las muestras pueden identificarse como los datos de difracción estándar de la estructura de perovskita distorsionada romboédrica correspondiente (JCPDS Card File No. 20 -0169, grupo espacial: R3c ). También se puede ver que todas las muestras exhiben patrones de difracción nítidos con una pequeña cantidad de fases secundarias. Se pueden ver rastros de una fase secundaria a 27,6 ° y 32,8 ° (marcado con "*" para Bi 2 Fe 4 O 9 y "#" para Bi 25 FeO 40 ) para x =0.5 y x =0,1 muestras que pueden resultar de la naturaleza de volatilización del Bi a la alta temperatura de sinterización [34]. Esto se observa a menudo en los polvos de BFO sintetizados por diferentes rutas [35,36,37]. Se encuentra que las fases secundarias aumentan continuamente a un alto valor de dopaje. Por lo tanto, la sustitución de Fe por Al en el sitio B no puede promover la fase pura de BFO; sin embargo, las propiedades eléctricas de las muestras no se verían afectadas. En la Fig. 1a, se puede ver que todos los picos de difracción para las muestras dopadas primero se desplazan a 2 θ superiores valores con dopaje de Al. Para mayor claridad, una parte de los patrones XRD en el 2 θ La región de 21 a 24 ° se amplifica en la Fig. 1b. Desde esta vista ampliada de los picos de difracción, se puede observar que el pico de difracción (101) tiene un desplazamiento obvio hacia 2 θ valores con respecto al BFO sin dopar, lo que confirma que Al se dopa con éxito en el sitio B del BFO. En analogía con el BFO sin dopar, el pico de difracción (101) de las muestras dopadas sufre un cambio en 2 θ superiores valores primero y luego un pequeño cambio en 2 θ inferiores valores cuando x =0.1 (rodeado por una línea discontinua), como se muestra en la Fig. 1c. En la figura 1d se muestran los patrones de XRD magnificados en las proximidades de 37 a 40 °, 50 a 52 ° y 55 a 57 °. Hay algunos picos gemelos, a saber, (003) y (021), (113) y (211), (104) y (122), en el espectro XRD. Con el aumento del contenido de Al, la intensidad de estos picos primero aumenta y luego disminuye cuando x llega a 0,1. Como sabemos, la intensidad de los picos suele estar relacionada con la cristalinidad. La disminución de los picos de difracción indica que la cristalinidad de BFA x O disminuye. Dado que Al 3+ (0,51 Å) tiene un radio iónico más pequeño que el Fe 3+ (0,65 Å), se incorpora fácilmente a la red de BFO cuando la cantidad de dopaje es pequeña, pero el exceso de iones de dopaje hace que la red de BFO sea inestable. La cristalinidad reducida puede deberse al hecho de que Al favorece la creación de más sitios de nucleación que, a su vez, inhiben el crecimiento de granos de cristal. También se ha encontrado una disminución de la cristalinidad en otros BFO dopados con Al [28, 30]. Por otro lado, esto puede ocurrir si se crean vacantes de oxígeno y se transforma algo de Fe 3+ a Fe 2+ debido al desequilibrio de carga creado en el sistema por Al 3+ sustitución. Se ha observado un fenómeno similar en BFO dopado con Sr [38]. El cambio en los picos de difracción podría atribuirse a la contracción de la celda unitaria, debido al pequeño radio iónico del Al en comparación con el Fe 3+ . Los resultados de XRD muestran que el trivalente Al 3+ La sustitución en BFO no conduce a una transformación estructural observable.
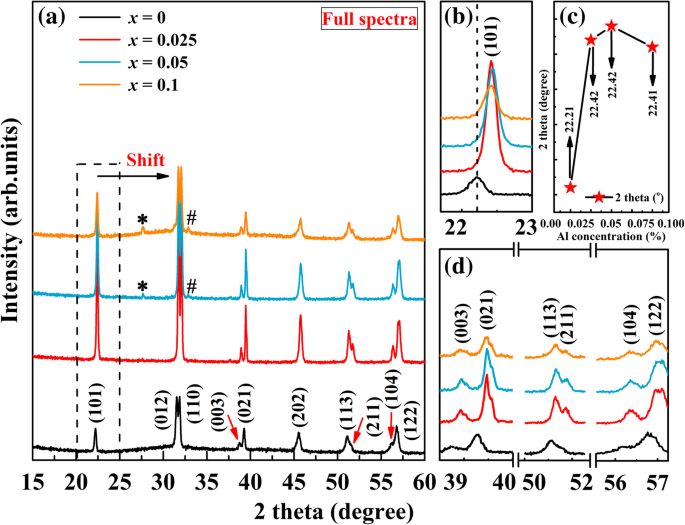
un Patrones XRD de BFA x O (0 ≤ x ≤ 0,1). b Vista ampliada de los patrones de XRD en el rango de 21–24 °. c La posición del pico (101) en función de la concentración de Al. d Patrones de XRD ampliados en las proximidades de 37–40 °, 50–52 ° y 55–57 °
La estructura confirmada por XRD también se puede caracterizar por la posición y la intensidad de los modos activos Raman. El espectro Raman es sensible al desplazamiento y distribución atómicos. Los espectros de dispersión Raman del BFO y BFA sin dopar x Los polvos O se muestran en la Fig. 2. Sobre la base de la teoría de grupos, los 13 modos activos Raman de fonón óptico y los 5 modos inactivos (es decir, 5 A 2 ) de BFO se predice para las perovskitas de distorsión romboédrica con el grupo espacial R3c [39]. El análisis teórico de las vibraciones reticulares en la perovskita R3c la estructura es la siguiente:
$$ {\ varGamma} _ {R3c} =4 {A} _1 \ left (z, \ kern0.5em {x} ^ 2, {y} ^ 2, \ kern0.5em {z} ^ 2 \ right) + 5 {A} _2 \ left (- \ right) + 9E \ left (x, \ kern0.5em y, \ kern0.5em {x} ^ 2- {y} ^ 2, \ kern0.5em xy, \ kern0. 5em xz, \ kern0.5em y \ right) $$ (1)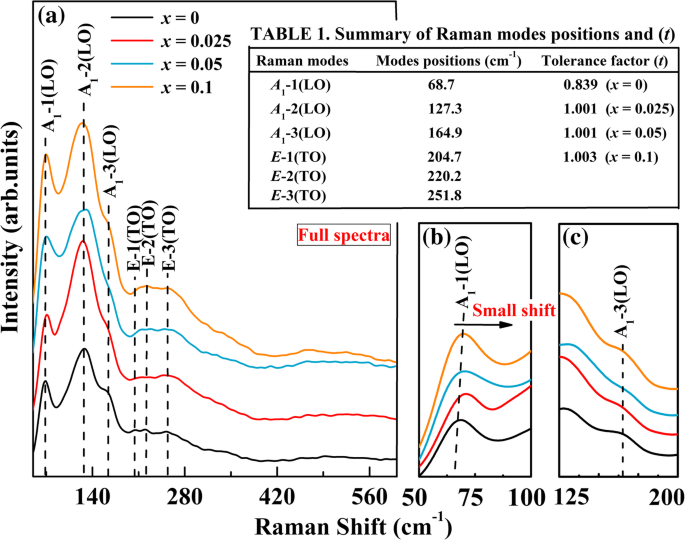
un Espectro Raman de BFA x O (0 ≤ x ≤ 0,1). b Espectro Raman ampliado en rangos de 50 a 100 cm −1 y c 125–200 cm −1
donde A es el modo óptico longitudinal (LO) y E es el modo óptico transversal (TO). En los espectros completos que se muestran en la Fig. 2a, el número de modos activos Raman claramente visibles en RT es mucho menor que el previsto. En este trabajo, hemos observado seis modos activos Raman (3 A 1 (LO) + 3 E (TO)) para BFA x Oh polvos. Esto puede deberse a degeneraciones accidentales entre bandas en los espectros y la incapacidad para distinguir bandas débiles del ruido de fondo [40], o fugas dieléctricas en la muestra. Para el BFO sin dopar, los picos fuertes y anchos a 68,7 cm −1 , 127,3 cm −1 y 164,9 cm −1 fueron asignados a A 1 -1 (LO), A 1 -2 (LO) y A 1 -3 modos (LO), respectivamente. Picos a 204,7 cm −1 , 220,2 cm −1 y 251,8 cm −1 fueron asignados a E -1 (TO), E -2 (TO) y E -3 (TO) modos, respectivamente (consulte la Tabla 1 en la Fig. 2). De los espectros, se puede ver claramente que estos E Los modos (TO) son invisibles. Se sabe que A 1 Los modos -1 (LO) se atribuyen a los enlaces Bi-O, mientras que la inclinación A 1 Los modos -3 (LO) se ven como FeO 6 octaedro. E Los modos (TO) se asignan a la vibración Fe-O [41]. El resultado de Raman confirma que el BFO sin dopar preparado pertenece a la estructura romboédrica de perovskita distorsionada con el grupo espacial R3c . Se observa que las cuatro muestras muestran patrones Raman y modos de vibración similares. Esto indicó el mismo romboédrico R3c grupo espacial, pero sus intensidades y frecuencias eran algo diferentes. En la vista completa del patrón Raman de BFA x O muestras, la posición máxima de A 1 Los modos -1 (LO) se desplazan ligeramente a una frecuencia más alta y los picos de A 1 Los modos -3 (LO) se ampliaron, lo que indica que el dopante Al va al sitio B de BFO. Las intensidades del E -1 (TO), E -2 (TO) y E Se encuentra que los modos -3 (TO) aumentan ligeramente para las muestras dopadas. Para mayor claridad, la A 1 -1 (LO) y A 1 -3 modos (LO) en la región de 50-100 cm −1 y 125–200 cm −1 se muestran en la Fig. 2b y c. De este espectro ampliado, está claro que A 1 Los modos -1 (LO) exhiben un pequeño cambio a frecuencias más altas al aumentar el contenido de Al. En comparación con los del BFO sin dopar, los picos de A 1 Los modos -3 (LO) se ampliaron para las muestras dopadas. El pequeño cambio de A 1 -1 modos (LO), leve ampliación de A 1 -3 (LO) modos y cambio de las intensidades de algunos E Los modos (TO) pueden estar relacionados con el cambio de los enlaces covalentes Bi-O y Fe-O y un esfuerzo de compresión en la muestra dopada con Al [42, 36]. Por otro lado, es posible que todos los cambios anteriores se puedan deber al hecho de que el sitio B Fe 3+ Los iones han sido sustituidos parcialmente por Al 3+ iones. Aparentemente, estos resultados Raman son consistentes con las observaciones de XRD. El factor de tolerancia de Goldschmidt ( t ) se utiliza ampliamente para evaluar la estabilidad geométrica y la distorsión de estructuras cristalinas [43], donde t se define por la relación de tres tipos de radios iónicos, como sigue:
$$ t =\ frac {\ left ({r} _ {\ mathrm {A}} + {r} _ {\ mathrm {O}} \ right)} {\ sqrt {2} \ \ left ({r} _ {\ mathrm {B}} + {r} _ {\ mathrm {O}} \ derecha)} $$ (2)donde r A es el radio de Bi 3+ , r B es el radio promedio de Fe 3+ y Al, y r O es el radio de O 2- . Sin embargo, los radios de Al y Fe 3+ son 0,51 Å y 0,65 Å, mientras que Bi 3+ y O 2- tienen los radios de 1.03 Å (según Ref. [44]) y 1.38 Å, respectivamente. La t valores para nuestros compuestos de perovskita estudiados BFA x Se encuentra que O son 0.839, 1.001, 1.001 y 1.003 para x =0, 0.025, 0.05 y 0.1, respectivamente (consulte la Tabla 1 en la Fig.2). El ABO 3 ideal Los compuestos adoptan una estructura compacta cúbica cuando el valor de t es 1, mientras que cuando t <1 o> 1, surge una deformación geométrica [45, 46]. A medida que aumenta la concentración de Al, el radio iónico promedio del sitio B disminuye, lo que conduce a un aumento adicional en la t valor de 0,839 a 1,003. Esto puede deberse al pequeño cambio en un estado simétrico bajo de BFO. Es bien sabido que el catión del sitio B en la perovskita está rodeado por seis aniones de oxígeno y, cuando se reemplaza por iones más pequeños, la distancia de coordinación disminuirá. Por tanto, para analizar claramente los efectos de la sustitución de Al, es necesario estudiar la estructura electrónica local del BFA x O muestras.
XAFS se divide en dos tipos, es decir, absorción de rayos X cerca de la estructura del borde (XANES) y estructura fina de absorción de rayos X extendida (EXAFS). En esta medida, cuando el rayo X penetra a través de la losa con una distancia x , la intensidad del haz de rayos X se reducirá a I = yo o e - μ x . XAFS mide la absorción de rayos X en función de la energía de los rayos X E , es decir, el coeficiente de absorción de rayos X μ ( E ) =- d En yo / dx se determina a partir de la disminución de la intensidad del haz de rayos X I con distancia x [47]. La proporción de I / Yo o se traza en función de E por encima de los umbrales Fe K -edge (7112 eV) y Bi L 3 (13,419 eV), que puede proporcionar información importante sobre la forma de la μ ( E ). Las transiciones electrónicas deberán seguir la regla de selección de dipolos. Una absorbancia de rayos X μ ( ω ) se puede obtener según la regla de oro de Fermi, de la siguiente manera:
$$ \ upmu \ left (\ omega \ right) \ propto \ sum \ limits_f {\ left | \ left \ langle f \ left | D \ right | \ left.i \ right \ rangle \ right. \ right |} ^ 2 \ \ delta \ left \ langle {E} _i- {E} _f + \ omega \ right \ rangle $$ (3)donde | i ⟩ Es el estado inicial, | f ⟩ Es el estado final, D es el operador dipolo, E i es la energía de | i ⟩, E f es la energía de | f ⟩ Y ω es la frecuencia de los fotones. Las características de XANES contienen información útil que está asociada con la estructura electrónica del absorbedor y el entorno local de una estructura atómica. Función XANES χ ( E ) se define de la siguiente manera:
$$ \ chi (E) =\ kern0.5em \ left [\ frac {\ mu (E) - {\ mu} _ {\ mathrm {o}} (E)} {\ varDelta {\ upmu} _ {\ mathrm {o}}} \ right] $$ (4)donde μ o ( E ) es el fondo suave similar a un átomo y Δμ o es un factor de normalización que surge del aumento neto en el borde de absorción de fondo atómico total. La ecuación estándar. (4) se utilizó para la sustracción y eliminación de fondo durante el procesamiento de datos XANES. En primer lugar, se resta una función de pre-borde suave para eliminar el fondo de los instrumentos. En segundo lugar, μ ( E ) se normaliza y luego se resta una función de fondo suave posterior al borde de μ ( E ) para obtener el μ o ( E ). La Figura 3a muestra los espectros completos de Fe K -edge XANES de un BFO y BFA sin dopar x O polvos junto con el compuesto de referencia Fe 2 O 3 investigado en este trabajo, mientras que la Fig. 3b informa el espectro XANES en el rango de energía de 7100–7180 eV. En la Fig. 3b, se puede ver que la forma de todos los espectros y posiciones de los picos es similar entre sí. Los bordes de absorción se desplazan ligeramente hacia energías más bajas al aumentar la concentración de Al, lo que puede atribuirse al efecto de cambio químico. La energía del borde de absorción cambia a una energía más baja con el estado de carga decreciente, lo que sugiere una configuración de enlace compleja en el sitio B. Es bien sabido que el desplazamiento de los bordes podría usarse para obtener un estado de oxidación medio. Las energías del borde de absorción para nuestras muestras estudiadas BFA x O son 7124.95 eV, 7123.87 eV, 7123.84 eV y 7123.80 eV para x =0, 0.025, 0.05 y 0.1, respectivamente. Para el compuesto de referencia Fe 2 O 3 , la energía del borde de absorción es 7126,14 eV (consulte la Tabla 2 en la Fig. 3). El borde de absorción de las muestras dopadas es más bajo que el del Fe 2 O 3 (Fe 3+ ) compuesto de referencia. Aumentando x Los valores conducen al cambio gradual del borde de absorción hacia el de FeO (Fe 2+ ) [48]. De esto, podemos notar que BFA x O es un valente mixto (Fe 3+ / Fe 2+ ) sistema. En otro aspecto, se observan tres características principales, el pico anterior al borde A1 y los picos posteriores al borde A2 y A3, en las cuatro muestras. El pico previo al borde A1, típico de BFO, corresponde a la transición prohibida por cuadrupolo eléctrico desde O 1 s nivel a Fe 3 d unos, con una pequeña mezcla de Al 3 d estados. A medida que aumenta la concentración de Al, la intensidad del pico anterior al borde A1 exhibe un pequeño aumento para las muestras dopadas (ver Fig. 3c). El pico post-borde A2 se atribuye al O 2 p transferencia de banda al Fe 3 d órbita, el llamado proceso de transferencia de carga de ligando a metal [49], mientras que el pico A3 es causado por los 1 s a 4 p transición con dipolo permitido [50]. Con el aumento de la concentración de Al, se puede ver claramente que la intensidad de estos dos picos posteriores al borde aumenta para las muestras dopadas (ver Fig. 3d). Todos los cambios anteriores en la intensidad máxima antes y después del borde se pueden entender en términos de competencia entre la hibridación de Fe 3 d y Al 3 d con O 2 p orbitales. Además, después del dopaje con iones Al, hay más 4 p desocupados orbitales que ocurren en BFA x O. Excepto estos, todos los espectros no muestran cambios significativos y estos resultados demuestran que los iones Al están parcialmente dopados en el sitio B de BFA x O.
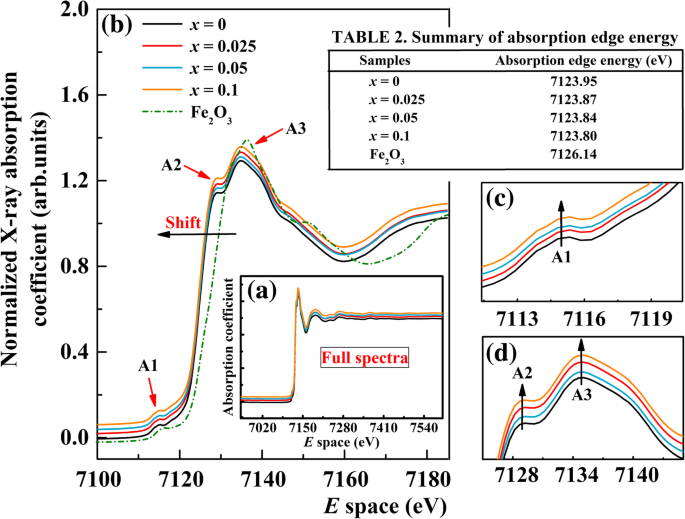
un Fe K espectros XANES de borde de BFA x O (0 ≤ x ≤ 0,1) y referencia Fe 2 O 3 . b Espectro XANES en el rango de 7100–7180 eV. c Vista ampliada del pico A. d Pico B y pico C
Las distribuciones de enlaces Fe-O, Fe-Fe / Al (es decir, Fe-O-Fe / Al) y Bi-O se obtienen ajustando la k 3 ponderado ( k 3 × χ ( k )) datos sin procesar, de la siguiente manera:
$$ k =\ sqrt {\ frac {2m \ left (E- {E} _ {\ mathrm {o}} \ right)} {\ mathrm {\ hbar}}} $$ (5)donde E o es la energía del borde de absorción y ħ es la constante de Planck. El ajuste adicional (algunas aproximaciones y oscilaciones) se realiza utilizando la ecuación estándar EXAFS, de la siguiente manera:
$$ \ chi (k) =\ sum \ limits_R {S} _ {\ mathrm {o}} ^ 2 {N} _ {\ mathrm {R}} \ frac {\ left | f (k) \ right |} {k {R} ^ 2} \ sin \ left (2 kR + 2 {\ delta} _ {\ mathrm {c}} + \ phi \ right) {e} ^ {\ frac {-2R} {\ lambda ( k)}} {e} ^ {- 2 {\ sigma} ^ 2 {k} ^ 2} $$ (6)donde \ ({S} _ {\ mathrm {o}} ^ 2 \) (\ (0 <{S} _ {\ mathrm {o}} ^ 2 <1 \)) es el factor de reducción, N R es el número de átomos de retrodispersión a una distancia R , f ( k ) es la amplitud de retrodispersión, δ c es el cambio de fase desde central, ϕ son los átomos de retrodispersión, λ es la vida útil del núcleo del agujero, σ 2 es el factor Debye-Waller desde múltiples distancias, y k es el camino libre medio de los fotoelectrones. La región EXAFS generalmente se refiere al rango de energía de 20 a 30 eV por encima del salto de borde, que es sensible a tipos de orden de corto alcance, distancias de enlace y números de coordinación en materiales. Aunque, Eq. (6) podría dar alguna información sobre las aproximaciones y oscilaciones de EXAFS, no es una forma particularmente conveniente para visualizar el contenido de información de un espectro de EXAFS. Por lo tanto, la transformación de Fourier se puede utilizar para descomponer un k señal espacial en sus diferentes frecuencias constituyentes [32]. La transformación de Fourier es una función compleja de la distancia interatómica R , cuya amplitud está representada por la función real de χ ( R ). En esta función, la posición de los picos está relacionada con las distancias de enlace y los iones vecinos. La ecuación (6) se puede transformar de k espacio para R espacio por transformada de Fourier, como sigue:
$$ \ chi (R) =\ frac {1} {\ sqrt {2 \ pi}} {\ int} _ {k _ {\ mathrm {min}}} ^ {k _ {\ mathrm {max}}} \ omega (k) {k} ^ n \ chi {e} ^ {- 2 ikR} dk $$ (7) $$ \ left \ {\ begin {array} {c} \ genfrac {} {} {0pt} {} {\ kern2.5em 0 \ kern7.5em k <{k} _ {\ mathrm {min}} \} {\ sin ^ 2 \ left [\ frac {\ pi \ left (k- {k} _ {\ mathrm {min}} \ right)} {2 \ left ({k} _2- {k} _ {\ mathrm {min}} \ right)} \ right] \ kern3.25em {k} _ {\ mathrm {min} }donde k máx y k min son los valores máximo y mínimo de k transformado espacio, respectivamente, χ (R) es la función de la ventana de Hanning, ω ( k ) es la función de ventana gaussiana, y k n es el factor de peso ( n =0, 1, 2, 3). El k 2 y k 3 valores para el BFA x O son 2 y 10, respectivamente, y el valor de n es 2. Transformada de Fourier de Fe K -edge EXAFS del BFA x Se realizan muestras O, como se muestra en la Fig. 4a. El pico asimétrico (rodeado por la primera línea discontinua) centrado en ~ 1.503 Å se identifica como un enlace Fe-O, debido a la dispersión de los aniones de oxígeno. El segundo pico fuerte (rodeado por la segunda línea de trazos) alrededor de ~ 3.527 Å corresponde a los enlaces Fe-Fe / Al, lo que puede explicarse por la dispersión de aniones de oxígeno de la vecina Fe / Al atómica próxima a la más cercana. conchas. La distancia para la primera y segunda capa de coordinación se ha resumido en la Tabla 3 en la Fig. 4. Desde la posición del pico, en comparación con la muestra de BFO sin dopar, el enlace Fe-O y Fe-Fe / Al tiende a desplazarse ligeramente hacia más pequeño R valores con x en aumento . Esto indica que el dopaje por iones Al no solo afecta a la estructura local del vecino más cercano del átomo de Fe central, sino que también afecta a las capas de coordinación más cercanas del átomo de Fe. Por otro lado, el cambio del enlace Fe-O a un R más pequeño Los valores pueden resultar de que el radio de los iones Al es menor que el de los iones Fe. Esto es consistente con los datos XRD. El cambio de Fe-Fe / Al a un R más pequeño Los valores también indican que la longitud promedio del enlace Fe-Fe / Al (donde los dos Fe 3+ los iones están en los centros del octaédrico de oxígeno vecino) gradualmente se acorta y el ángulo de enlace se modifica en las muestras dopadas con Al. However, there is a small increase of peak intensity of the Fe-O distribution for the doped samples while the peak intensity of the Fe-Fe/Al distribution is almost not changed. This reveals that the iron-neighboring structure of Fe-O has been modified by Al doping. The shorter Fe-Fe/Al bonds in Al-doped samples can explain why the major peaks (101) in XRD shift to higher 2θ angles (see Fig. 1b). This indicates that the substitution of Al for Fe could affect oxygen octahedral, which further reduces the coordination distance between the two neighboring Fe atoms. Figure 4b shows the Fe K -edge EXAFS of the BFAx O samples processed on k 3 × χ ( k ) oscillation with a k space of 0–10 Å −1 . As can be seen, all the k 3 × χ ( k ) spectra show similar patterns at the smaller k values but different at larger k values with some various noise (surrounded by the dash line). Al-doped samples show a broader k 2 × χ ( k ) spectrum than those of undoped BFO in the k space of 8.2–9.3 Å −1 , implying an enhanced short-range structural disorder in BFAx O samples. The noises are observed in a k space of 10–10.4 Å −1 . These changes indicated that the local structure of center atoms has changed due to B-site Al doping, similar to what was reported by Li et al. [51].

un Fourier transforms of Fe K -edge k 3 -weighted EXAFS data, for the BFAx O (0 ≤ x ≤ 0.1). b EXAFS χ ( k ) × k 3 spectra of BFAx O (0 ≤ x ≤ 0.1). The spectra were aligned along the Y -axis for better comparison
The peak positions, intensities, and shapes of the line in the Bi L 3 -edge XANES spectrum are well known to depend on the local electronic structure of the Bi atoms, which could provide information on the Bi valence. The full spectra of Bi L 3 -edge XANES of undoped BFO and BFAx O samples are also shown in Fig. 5a, while Fig. 5b shows the Bi L 3 -edge XANES spectrum in the energy region of 13,400–13,480 eV. The analysis of these spectra helps to investigate the local electronic structure of Bi ions in the doped system. From Fig. 5b, it can be seen that the shape of all spectra are the same to each other and there is almost no change of absorption edge in the whole series. The absorption edge energies for our study are found to be 13,429.1 eV, 13,429.4 eV, 13,429.3 eV, 13,429.3 eV, and 13,429.8 eV for x =0, 0.025, 0.05, and 0.1 and the reference compound Bi2 O3 , respectively (see Table 4 in Fig. 5). The absorption edges slightly shift toward higher energies with increasing Al concentration. The absorption edge in the Bi L 3 -edge of the BFAx O samples matches well with that of the reference compound Bi2 O3 , which indicates that the valence state of Bi ions in all the samples is in + 3 valence state. However, there are two post-edge peaks found in all samples and marked as B1 and B2, respectively. These two post-edge peaks are caused by the electric-forbidden transition from 2p 3/2 level to the 6d unos. Compared with undoped BFO, the intensity of peak B2 can be clearly seen to increase for the doped samples (see Fig. 5c), which means the transition from 2p 3/2 to 6d state increases, so does the energy of 6d state. Except these, there is no other significant change in the whole spectrum.

un Bi L 3 -edge XANES spectra of BFAx O (0 ≤ x ≤ 0.1) and reference Bi2 O3 . b XANES spectrum in the range of 13,400–13,480 eV. c Enlarged view of the peak D and peak E
Fourier transform of Bi L 3 -edge EXAFS radial distribution functions is performed, as shown in Fig. 6a. A high-intensity peak located at around 1.618 Å corresponds to the nearest Bi-O coordination shell (surrounded by the dash line), which is a result from scattering from the nearest-neighbor atomic shell of Bi, i.e., oxygen anions. However, the position of Bi-O bond shifts toward larger R values for the doped samples (see Table 5 in Fig. 6). This indicates that the substitution of Al for Fe could affect the nearest-neighbor local structure of the central Bi atom. It also indicates the extension of Bi-O bond length. The peak intensity of the Bi-O distribution exhibited a small increase with increasing Al content, which suggests that the iron-neighboring structure of Bi-O has changed. Figure 6b shows the Bi L 3 -edge k 3 × χ ( k ) EXAFS spectra with a k space of 0–14 Å −1 . From Fig. 6b, it can be seen that all the spectra shape shows similar patterns except some error noises. The error noises are observed in the k space of 12–14 Å −1 (surrounded by the dash line). This result may imply that the k 3 × χ ( k ) EXAFS function of the center Bi atoms has changed with Al doping. This also suggests that the B-site Al substitution influences short-range structural disordering.

un Fourier transforms of Bi L 3 -edge k 3 -weighted EXAFS data, for the BFAx O (0 ≤ x ≤ 0.1). b EXAFS χ ( k ) × k 3 spectra of BFAx O (0 ≤ x ≤ 0.1). The spectra were aligned along the Y -axis
The optical properties of the samples are studied by using RT UV-Vis, which is used to characterize the optical properties of the materials. The UV-Vis absorption spectra of undoped BFO and BFAx O samples in the wavelength of 300–800 nm are shown in Fig. 7a. As a result, the UV-Vis spectra of the undoped BFO and BFAx O samples show two absorption edges (marked by dashed arrows). One is a band around 650 nm, which is due to the metal-to-metal transition. The other is a band around 760 nm, which is caused by crystal field transition [52]. In addition, the strong absorption band is observed at about 490 nm (marked by dashed arrow), which is attributed to the electronic transition from O 2p to Fe 3d state in the BFO. These strong bands indicate that the BFO prepared by hydrothermal method could be a promising visible-light photocatalytic material. BFO is of direct transition with a value of n as 2. The absorption edge of the doped samples shifted from 659 to 619 nm, suggesting that the BFAx O powders absorb visible light in the wavelength range of 600–659 nm (see Fig. 7a). A similar blue-shift phenomenon was observed earlier in other element-doped BFO [53,54,55]. This blue shift in the absorption spectra of Al-doped samples in comparison with the undoped BFO shows that doping Al causes a change in the local structure for BFO. From Fig. 7a, one can see that the absorption spectra of the Al-doped samples exhibit a sharp increase around 490 nm and it suggests that all samples can absorb remarkable amounts of visible light. For the sample with x =0.025, the absorption spectrum shows a sudden increase. It means that it has a wider absorption range than the other samples in this range of visible light. The optical band gap of the samples has been calculated by Tauc’s formula, as follows:
$$ ahm=A{\left( hm-E\mathrm{g}\right)}^n $$ (9)
un UV-Vis absorption spectrum of BFAx O (0 ≤ x ≤ 0.1). b Plots of (ahν ) 2 vs. photon energy. c E g values as a function of Al concentration
where a is the absorption coefficient, A is the parameter, h is the Planck’s constant, m is the frequency of the incident photon, E g is the optical band gap, and n (for direct n =2, for indirect n =0.5) is a constant associated with different types of electronic transitions, as shown in Fig. 7b. The calculated E g values are found to be 1.833 eV, 1.888 eV, 1.866 eV, and 1.905 eV for x =0, x =0.025, x =0.05, and x =0.1 samples, respectively. It is easy to see that the band gap increases with the substitution ratio, as shown in Fig. 7c. The increase in the band gap is attributed to the doping effect. The E g value for the undoped BFO is about 1.833 eV, which is lower than the previous reports [56, 57].
Conclusion
In summary, the BFAx O (x =0, 0.025, 0.05, and 0.1) multiferroic powder samples were successfully synthesized via hydrothermal route. Effects of Al substitution on the structural, electrical, and optical properties of the samples were studied. The structural study reveals that Al-doped BiFeO3 shows the existence of secondary phases and lattice contraction due to lower ionic radii of Al doped into B-site, which still retains its rhombohedral R3c perovskite structure. Raman scattering measurement infers six Raman active phonon modes, which further confirms the result of XRD. XAFS studies on the Fe K -edge and B L 3 -edge of the BFAx O samples and of the reference compounds Fe2 O3 y Bi 2 O3 were performed, and the obtained results were compared in order to determine the valance states of Fe and Bi ions in the system. The Fe K -edge XAFS results revealed that BFAx O is a mixed-valent (Fe 3+ /Fe 2+ ) system. The results of Fe K -edge XAFS also illustrate a competition between the Fe 3d and Al 3d orbitals on hybridization with the O 2p and occurrence of the more 4p orbitals with Al doping. Besides, Al ion doping affects both the nearest-neighbor and next-nearest coordination shells of the Fe atom. The B L 3 -edge XAFS results indicate that valence states of Bi ions in all the samples are in + 3 and the transition from 2p 3/2 to 6d state and the energy of 6d state increases. Substitution Al for Fe could affect the nearest-neighbor local structure of central Bi atom. The BFAx O prepared by hydrothermal method could be an appropriate visible-light photocatalytic material due to a strong absorption band in the visible region.
Abreviaturas
- AFM:
-
Antiferromagnetic
- BFAx O:
-
BiFe1-x Alx O3
- BFO:
-
BiFeO3
- EXAFS:
-
X-ray absorption fine structure
- FE:
-
Ferroelectric
- FM:
-
Ferromagnetic
- RT:
-
Temperatura ambiente
- UV-Vis:
-
Ultraviolet-visible
- XAFS:
-
X-ray absorption fine structure
- XANES:
-
X-ray absorption near edge structure
- XRD:
-
Difracción de rayos X
Nanomateriales
- Absorción de luz mejorada con plasmón en (p-i-n) células solares de nanocables GaAs de unión:un estudio de método de simulación FDTD
- Expansión térmica anómala de HoCo0.5Cr0.5O3 sondeado por difracción de polvo sincrotrón de rayos X
- El estudio de un nuevo sistema micelar similar a un gusano mejorado con nanopartículas
- Investigación sobre la polarización de la superficie de la heteroestructura de GaN / AlGaN / GaN con cubierta de Al2O3 mediante espectroscopia de fotoelectrones de rayos X de ángulo resuelto
- Estudio antitumoral de nanogeles de condroitina sulfato-metotrexato
- Mejora de la absorción y modulación de frecuencia del microbolómetro THz con estructura de micropuente mediante antenas de tipo espiral
- Estructura electrónica y características I-V de las nanocintas InSe
- Propiedades ópticas de películas de ZnO dopado con Al en la región infrarroja y sus aplicaciones de absorción
- Estudio de los primeros principios de los defectos puntuales en la superrejilla de GaAs / AlAs:la estabilidad de fase y los efectos sobre la estructura de la banda y la movilidad del portador
- Adsorción de metales de transición en fosforeno negro:un estudio de los primeros principios
- Espectroscopia de absorción mejorada por cavidad (CEAS) para la detección de ozono