


Adsorción de metales de transición en fosforeno negro:un estudio de los primeros principios
Resumen
El fosforeno negro es un material bidimensional novedoso que tiene propiedades únicas y amplias aplicaciones. Utilizando cálculos de primeros principios, investigamos el comportamiento de adsorción de 12 metales de transición diferentes (TM; Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt y Au) en fosforeno. Nuestros resultados mostraron que todos los sistemas de adsorción tienen una gran energía de enlace. Los sistemas de Fe-, Co- y Au-fosforeno muestran estados magnéticos con momentos magnéticos de 2, 1 y 0,96 μ B , respectivamente, lo que significa que estos sistemas son semiconductores magnéticos. También se investigó la adsorción de moléculas de oxígeno en TM-fosforeno. Curiosamente, todos los O 2 - Sistemas (TM-fosforeno), excepto O 2 - (Pd-fosforeno), puede alargar el enlace O – O, que es fundamental para su aplicación como catalizadores en la oxidación de CO. También encontramos que la adsorción de O 2 moléculas habilita el O 2 - Los sistemas (Fe-, Ni-, Cu-, Ir-, Rh-, Ag- y Au-phosphorene) se convierten en semiconductores magnéticos, y permite que O 2 - (Co-fosforeno) para mostrar el estado semimetálico. Se espera que nuestros resultados tengan implicaciones importantes para la catálisis basada en fosforeno y la espintrónica.
Antecedentes
El fosforeno [1,2,3], una monocapa de átomos de fósforo dispuestos en una estructura de panal arrugada, tiene propiedades únicas que incluyen una naturaleza semiconductora directa [4], movilidad ultra alta a temperatura ambiente [4,5,6], flexibilidad mecánica superior [7] y alto rendimiento termoeléctrico [8,9,10]. Estas propiedades hacen que el fosforeno sea un material muy adecuado para una variedad de aplicaciones, como transistores de efecto de campo [1, 11,12,13,14,15,16], baterías de iones de litio y sodio [17,18,19], células solares [20, 21], fotocatalizadores [22], espintrónica [23] y sensores de gas [24, 25, 26]. Sin embargo, el fosforeno es un material no magnético y se deben adoptar algunas estrategias para ampliar su aplicación.
Para materiales bidimensionales (2D), la adsorción generalmente se selecciona como el enfoque para inducir el magnetismo para aplicaciones específicas. Previamente, Cao et al. [27] mostró que las propiedades electrónicas y magnéticas del grafeno pueden ser moduladas eficazmente por adatomos de Fe, Co, Ni y Cu. Kaloni y col. [28] demostró que los momentos magnéticos pueden ser inducidos en sistemas de siliceno decorados con Ti-, V-, Cr-, Mn-, Fe- y Co-decorados usando cálculos de primeros principios. Ersan y col. [29] encontró que b -Arsenene mostró caracteres de espín polarizados después de la adsorción de átomos de H, B, C, P, Ge, As y Sb. Además, w -Arsenene puede alcanzar momentos magnéticos netos con los adatomos de H, B, N, P, Cl, Ti, As y Sb. Para el fosforeno negro, Kulish et al. [30] predijo que Ag-, Au-, Ti-, V-, Cr-, Mn-, Fe- y Co-phosphorene son bastante estables, y una diversa gama de momentos magnéticos pueden ser inducidos en cálculos teóricos. Además, las propiedades de diferentes tipos de portadores de carga también se pueden ajustar adsorbiendo diferentes átomos en fosforeno. Ding y Wang [31] utilizaron los cálculos de los primeros principios para ilustrar sistemáticamente las propiedades estructurales, electrónicas y magnéticas de los átomos adsorbidos en fosforeno. Observaron que los adatomos pueden introducir magnetismo en el fosforeno, y los adatomos de P, Co y Au inducen propiedades magnéticas estables. Hu y Hong [32] utilizaron los cálculos de los primeros principios para demostrar las propiedades magnéticas de los adatomos metálicos sobre el fosforeno; demostraron que el magnetismo se puede obtener en el fosforeno adsorbiendo átomos de Cr, Fe, Co o Au en su superficie. Además, predijeron que el sistema de adsorción de Fe-fosforeno será un material semiconductor magnético diluido prometedor. Por lo tanto, se puede esperar que la adsorción de metales de transición (TM) en el fosforeno negro sintonice de manera efectiva las propiedades magnéticas del material.
Aunque las investigaciones anteriores estudiaron el comportamiento de adsorción de los metales de transición en el fosforeno negro, algunos problemas siguen sin resolverse. Por ejemplo, los estudios anteriores se centraron principalmente en las propiedades de las 3d TM adsorbidas en fosforeno. ¿Cómo diseñarán las TM 4d y 5d las propiedades del fosforeno? Además, los metales nobles absorbidos en fosforeno también se pueden utilizar como catalizadores de un solo átomo. Li y col. [33] sugirió que el siliceno con Au adsorbido puede ser un catalizador de alta actividad con barreras de baja energía catalítica para la oxidación del CO. ¿Puede un metal noble absorbido en fosforeno también ser un buen candidato para la oxidación del CO? Para responder a estas preguntas, presentamos en este artículo los resultados de un estudio detallado de los primeros principios sobre las propiedades estructurales, magnéticas y electrónicas de 12 tipos diferentes de átomos de metales de transición adsorbidos en el fosforeno negro. Seleccionamos Fe, Co y Ni elementales, que son metales ferromagnéticos en su fase a granel; Cu elemental, que es diamagnético; y los metales nobles Ru, Rh, Pd, Ag, Os, Ir, Pt y Au, que son muy eficaces para la oxidación del CO [19, 34,35,36,37,38,39,40,41,42 , 43, 44, 45]. Descubrimos que el fosforeno forma enlaces fuertes con los 12 metales y que todos los sistemas de TM-fosforeno son bastante robustos. Las propiedades electrónicas y magnéticas del fosforeno pueden ajustarse eficazmente mediante los adatomos. Además, también encontramos que la mayoría de los sistemas de adsorción de TM-fosforeno son buenos candidatos para el catalizador en la oxidación del CO. Los resultados de esta investigación se pueden utilizar para estudios fundamentales del fosforeno y también pueden ampliar su aplicación potencial en muchos campos importantes. .
Métodos / Experimental
Nuestros cálculos se basaron en la teoría funcional de densidad polarizada por espín (DFT), y se realizaron utilizando el paquete de simulación Vienna Ab Initio (VASP) [46, 47] y la aproximación de gradiente generalizada (GGA) de Perdew-Burke-Ernzerhof ( PBE) funcional [48,49,50]. Se utilizó el método DFT-D3 de Grimme [51] para calcular la interacción de van der Waals. Se empleó un corte de energía de 400 eV con un conjunto de base de onda plana. En los cálculos, los átomos se relajaron hasta que la energía total convergió a 1 × 10 −5 eV y la fuerza residual en cada átomo fue inferior a 0,01 eV / Å. Se usó una gran supercélula (4 × 3) a lo largo de las direcciones en zigzag y del sillón para evitar interacciones entre las celdas unitarias vecinas. Las constantes de celosía se establecieron en a =13,20 Å y b =13,74 Å. Aplicamos un espacio de vacío de 20 Å en la z dirección para minimizar las interacciones entre capas intermedias adyacentes. Durante la optimización, un Monkhorst-Pack [52] k -se adoptó una cuadrícula de 3 × 3 × 1, y una k Se utilizó una cuadrícula de puntos de 7 × 7 × 1 para los cálculos de energía total.
Resultados y discusión
Primero exploramos las propiedades estructurales del fosforeno prístino. La figura 1a muestra las ilustraciones de las vistas superior y lateral de la estructura cristalina. Puede verse que la monocapa de fosforeno consta de dos planos atómicos y la celda unitaria de fosforeno consta de cuatro átomos de P. La monocapa de fosforeno tiene una red tetragonal con constantes de red de equilibrio a =3.30 Å y b =4,58 Å. La longitud del enlace P – P en la dirección horizontal ( l 1 ) es 2,22 Å, mientras que la longitud en la otra dirección ( l 2 ) es 2,26 Å. El fosforeno prístino tiene una banda prohibida directa de 0.89 eV (Fig. 1b), con el mínimo de la banda de conducción (CBM) y el máximo de la banda de valencia (VBM) ubicados en el punto Г. La constante de celosía y la banda prohibida que obtuvimos están muy de acuerdo con los valores obtenidos en estudios de investigación previos [30, 31, 32, 53].
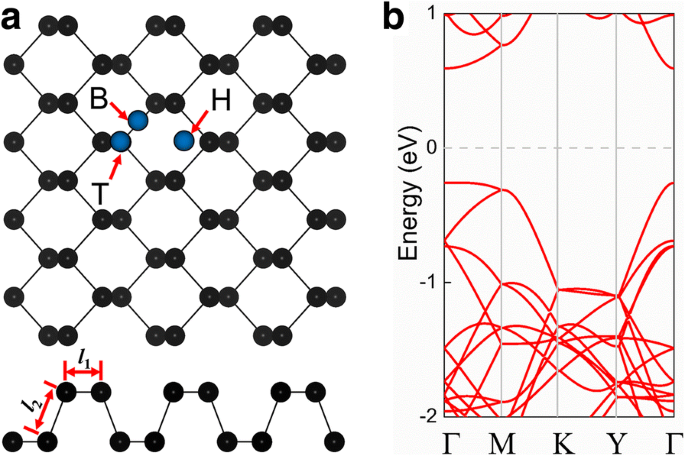
un Vistas superior y lateral de la estructura cristalina de fosforeno prístino (supercélula 4 × 3 × 1). Los círculos azules representan posiciones típicas de un átomo de impureza adsorbido en un punto hueco (H), en un puente (B) entre dos átomos de fósforo y encima de un átomo de fósforo (T). b Estructura de banda electrónica y primera zona Brillouin de fosforeno prístino; el nivel de Fermi se establece en cero
Un adatom típico siempre se adsorbe en una de estas tres posiciones:encima de un sitio hueco (H), en un puente (B) entre dos átomos de fósforo y encima de un átomo de fósforo (T). Calculamos la energía de adsorción de un adatom sobre fosforeno para examinar la estabilidad de los sistemas de adsorción usando la relación:
$$ {E} _ {\ mathrm {ad}} =\ left ({E} _ {\ mathrm {TM}} + {E} _ {\ mathrm {fosforeno}} \ right) - {E} _ {\ mathrm {TM} - \ mathrm {fosforeno}} $$ (1)donde E TM es la energía de un átomo metálico aislado, E fosforeno es la energía total de la capa prístina de fosforeno, y E TM-fosforeno es la energía total del sistema de adsorción. Según esta ecuación, una mayor energía de adsorción indica una estructura más estable. Descubrimos que todos los átomos de metal estudiados en nuestro trabajo prefieren permanecer en el sitio H del fosforeno. Las energías de adsorción calculadas de los átomos de metal adsorbidos en el sitio H del fosforeno, que se muestran en la Tabla 1, varían de 2 a 6 eV. La longitud de enlace de TM-fosforeno ( d TM-P ) demostró ser corto, en el rango de 2,11-2,43 Å. El análisis de carga de Bader [54, 55, 56] muestra que 0,16, 0,16, 0,07, 0,17, 0,32, 0,33 y 0,16 | e | se transfieren de los átomos de metal Ru, Rh, Pd, Os, Ir, Pt y Au, respectivamente, a fosforeno en los sistemas de adsorción de (4d-TM) -fosforeno y (5d-TM) -fosforo. Todos estos resultados denotan la formación de enlaces químicos entre el TM adatom y el fosforeno. Además, estos resultados están cerca de estudios recientes [30,31,32].
Como se muestra en la Tabla 1, los sistemas Ni-, Cu-, Ru-, Rh-, Pd-, Ag-, Os-, Ir- y Pt-fosforeno exhiben estados no magnéticos, mientras que los sistemas Fe-, Co- y Au -los sistemas de fosforeno tienen momentos magnéticos de 2, 1 y 0,96 μ B , respectivamente. La densidad de carga polarizada por espín ( ρ = ρ giro - ρ spin-down ) también se muestra en la Fig.2 para explorar el origen y distribución del magnetismo en los sistemas de adsorción magnéticos de TM-fosforeno. El momento magnético en cada uno de estos casos se origina principalmente en el adatom, con un pequeño momento magnético resultante de los vecinos más cercanos. Además, las estructuras de bandas calculadas de los sistemas de Fe-, Co- y Au-fosforeno se muestran en la Fig. 2. Puede verse que estos sistemas son todos semiconductores magnéticos con bandgaps de 0.38, 0.22 y 0.06 eV, respectivamente. que son útiles para aplicaciones espintrónicas.
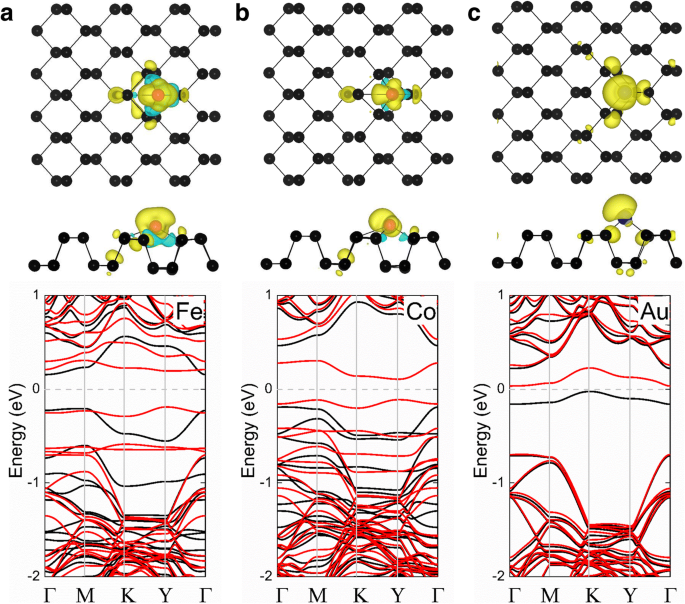
Densidades de centrifugado de a Fe-fosforeno, b Cofosforeno y c Los sistemas de au-fosforeno se muestran en la fila superior; la estructura de bandas correspondiente de cada sistema se muestra en la fila inferior. Las esferas negra y roja representan átomos de P y TM, respectivamente. En la fila superior, un gráfico de la densidad de carga polarizada por espín con un valor de densidad de carga iso-superficial de 0,002 e / Å 3 se superpone en las vistas superior y lateral de la estructura cristalina de fosforeno prístino para cada uno de los sistemas TM-fosforeno; las regiones amarilla y cian corresponden a los giros hacia arriba y hacia abajo, respectivamente. En el gráfico de estructuras de bandas (fila inferior), las líneas negra y roja indican canales de rotación hacia arriba y hacia abajo, respectivamente; el nivel de Fermi se establece en cero y se indica mediante la línea discontinua gris
A continuación, estudiamos el comportamiento de adsorción de O 2 encima del átomo de TM en los sistemas TM-fosforeno. Dos configuraciones típicas de menor energía para la adsorción de O 2 en sistemas TM-fosforeno (O 2 - (TM-fosforeno)) se muestran en la Fig. 3. Para O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Pd-fosforeno) y O 2 - Sistemas (Pt-fosforeno), el O 2 La molécula es paralela a la dirección en zigzag del fosforeno (Fig. 3a), con una longitud de enlace O – P de 1,84 Å, 1,86 Å, 2,04 Å, 2,18 Å y 2,05 Å, respectivamente. Para el O 2 - (Ni-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno) y O 2 - Sistemas (Au-fosforeno), la molécula se encuentra a lo largo de la dirección en zigzag del fosforeno (Fig. 3b), en un cierto ángulo desde la superficie. Mientras tanto, los dos átomos de O vecinos alrededor del adatom TM no son equivalentes. Los resultados se muestran en la Tabla 2. La energía de adsorción ( E anuncio ) de O 2 en un O 2 - El sistema (TM-fosforeno) se calculó como:
$$ {E} _ {\ mathrm {ad}} ={E} _ {\ mathrm {TM} - \ mathrm {fosforeno}} + {E} _ {{\ mathrm {O}} ^ 2} - {E } _ {{\ mathrm {O}} ^ 2- \ mathrm {TM} - \ mathrm {fosforeno}} $$ (2)donde \ ({E} _ {{\ mathrm {O}} ^ 2- \ mathrm {TM} - \ mathrm {fosforeno}} \), E TM-fosforeno , y \ ({E} _ {{\ mathrm {O}} ^ 2} \) son las energías totales de O 2 - Sistema (TM-fosforeno), el sistema TM-fosforeno y el O 2 molécula, respectivamente. Como se muestra en la Tabla 2, las energías de adsorción son 2.659, 1.850, 0.970, 0.906, 2.402, 1.548, 0.001, 0.786, 3.109, 1.980, 0.416 y 1.029 eV para O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Pd-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Pt-fosforeno) y O 2 - Sistemas (Au-fosforeno), respectivamente. En todos los casos, las grandes energías de adsorción excepto la del O 2 - El sistema (Pd-fosforeno) indica que O 2 es quimisorbido.
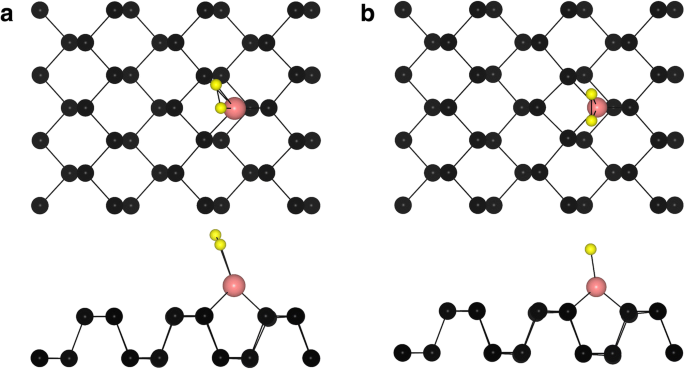
Vistas superior y lateral de los sitios de adsorción típicos de un O 2 molécula de TM-fosforeno. Las esferas negra, rosada y amarilla representan átomos de P, TM y O, respectivamente
Se reconoce bastante que el alargamiento del enlace O – O es crucial para los mecanismos de Langmuir-Hinshelwood y Eley-Rideal de un catalizador en la oxidación del CO [57]. En términos generales, cuanto más larga sea la longitud del enlace O – O, más fácil será la reacción del catalizador. Las longitudes de enlace O – O y TM – O en cada sistema también se muestran en la Tabla 2. Obviamente, el enlace O – O aumenta de 1.23 Å para el O 2 prístino molécula a 1,38, 1,36, 1,32, 1,35, 1,40, 1,34, 1,32, 1,30, 1,46, 1,39, 1,40 y 1,32 Å, respectivamente, para la molécula adsorbida, posiblemente porque O 2 es un aceptor de electrones. Además, la longitud de enlace de TM – O en la mayoría de O 2 - Los sistemas (TM-fosforeno) son cortos debido a la interacción entre O 2 y los átomos de TM. Esta longitud de enlace varía de 1,84 a 2,19 Å y da como resultado la formación de enlaces químicos. En particular, el enlace O – O se alarga a 1,40 Å, el valor más alto entre los sistemas, en el O 2 adsorbido. molécula en el sistema Pt-fosforeno. Por lo tanto, el sistema de Pt-fosforeno es bastante adecuado como catalizador para la oxidación de CO porque probablemente tiene una alta capacidad catalítica.
Para obtener más información sobre el mecanismo subyacente de la alta actividad de estos sistemas, seleccionamos O 2 - (Pt-fosforeno) como ejemplo e investigó su densidad local de estados (LDOS). La Figura 4a muestra el LDOS proyectado en los orbitales d de Pt en el sistema Pt-fosforeno, orbitales d de Pt en el O 2 - Sistema (Pt-fosforeno), el enlace O – O en el O 2 - Sistema (Pt-fosforeno) y la fase gaseosa O 2 . En el panel superior de la Fig. 4a, se puede ver un pico en E F - 0,6 eV, que se origina en el orbital d parcialmente ocupado de Pt en el sistema Pt-fosforeno. Estos estados deberían ser responsables de la alta actividad del sistema Pt-fosforeno. Después de la adsorción de un O 2 molécula, el LDOS proyectado en los orbitales d de Pt por debajo del nivel de Fermi se reduce después de la adsorción del O 2 molécula debido a la transferencia de carga, y los estados por encima del nivel de Fermi también aumentan sustancialmente. Mientras tanto, el LDOS se proyecta sobre el O 2 adsorbido molécula indica que el O 2 2 π * Los orbitales (orbital molecular desocupado más bajo, LUMO) se están ocupando parcialmente, lo que se ha reducido de su valor de gas de E F + 2 eV a E F - 0,1 eV. Para aclarar, la diferencia de densidad de carga del O 2 - También se presenta el sistema (Pt-fosforeno).
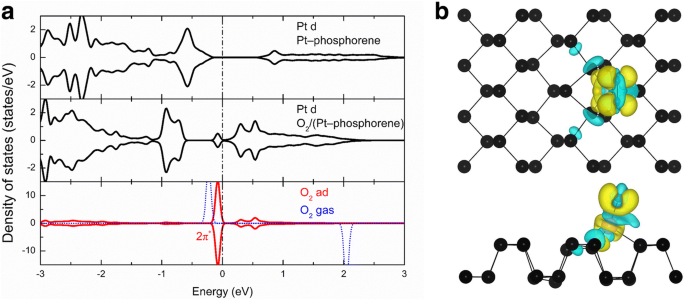
un Densidad local de estados (LDOS) de Pt y O 2 moléculas en Pt-fosforeno y O 2 -Sistemas de pt-fosforeno y fase gaseosa O 2 , respectivamente. b Diferencia de densidad de carga en el O 2 - Sistema (Pt-fosforeno); la región amarilla (es decir, + 0,002 e / Å 3 ) y la región cian (es decir, - 0,002 e / Å 3 ) corresponden al aumento y la pérdida, respectivamente, de la densidad electrónica
La diferencia de densidad de carga se define de la siguiente manera:
$$ {\ varDelta} _ {\ rho} ={\ rho} _T - {\ rho} _ {\ mathrm {molécula}} - {\ rho} _ {\ mathrm {absorbida}} $$ (3)donde ρ T , ρ molécula y ρ absorbido son los cargos totales en el O 2 - Sistema (Pt-fosforeno), O 2 molécula y el sistema Pt-fosforeno, respectivamente. Como se muestra en la Fig. 4b, la gran región amarilla localizada en el O 2 molécula indica que hay una transferencia de electrones significativa de Pt-fosforeno a O 2 , que también indica la fuerte hibridación orbital entre O 2 y el sistema Pt-fosforeno. Según el análisis de carga de Bader [54,55,56], 0,19 | e | se transfiere del sistema de Pt-fosforeno al O 2 molécula. Por lo tanto, la gran transferencia de carga llena los estados antienlazantes del O 2 molécula y debilita significativamente el enlace O – O. De manera similar, el mecanismo subyacente de la alta actividad de otros sistemas también puede entenderse por la transferencia de carga entre O 2 molécula y el sistema TM-fosforeno. De hecho, el análisis de carga de Bader [54,55,56] mostró que las cargas de - 0,68, - 0,50, - 0,42, - 0,52, - 0,46, - 0,24, - 0,24, - 0,37, - 0,53, - 0,25, - 0,19 y - 0,09 | e | se transfieren de TM-fosforeno a la molécula de oxígeno en el O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Pd-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Pt-fosforeno) y O 2 - Sistemas (Au-fosforeno), respectivamente.
Finalmente, estudiamos las propiedades magnéticas de O 2 - Sistemas (TM-fosforeno). Los momentos magnéticos del O 2 - Los sistemas (TM-fosforeno) se muestran en la Tabla 3. El O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno) y O 2 - Los sistemas (Ir-fosforeno) tienen momentos magnéticos de 2,00, 1,00, 1,00, 1,14 y 1,00 μ B , respectivamente, que resultan de la adsorción de un O 2 paramagnético molécula. La densidad de carga polarizada por espín de estos O 2 - Los sistemas (TM-fosforeno) se muestran en la Fig. 5. Para el O 2 - (Fe-fosforeno) y O 2 - (Co-fosforeno), se cree que el momento magnético surge principalmente del átomo del metal de transición y el O 2 molécula. Al contrario, para el O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Ir-fosforeno) y O 2 - Sistemas (Au-fosforeno), el momento magnético proviene principalmente del O 2 molécula. Estas hipótesis son consistentes con los resultados mostrados en la Tabla 3. Para comprender mejor cómo la adsorción de una molécula de gas afecta la estructura electrónica del O 2 - sistema (TM-fosforeno), se calcularon las estructuras de bandas electrónicas de cada sistema y los resultados se muestran en la Fig. 5. Primero, descubrimos que se produce una banda plana alrededor del nivel de Fermi ( E F ) después de la adsorción de O 2 molécula en todos los sistemas, que principalmente del O 2 molécula. Para el O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Ag-fosforeno) y O 2 - Sistemas (Au-fosforeno), los canales para la división del spin-up y spin-down revelan las características magnéticas. El O 2 - (Fe-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno) y O 2 - (Au-fosforeno) exhiben un comportamiento semiconductor magnético, con una banda prohibida considerable excepto para el O 2 - Sistema (Co-fosforeno), que resultó ser semimetálico. Estos resultados sugieren que los sistemas tienen potencial para su aplicación en espintrónica basada en fosforeno.
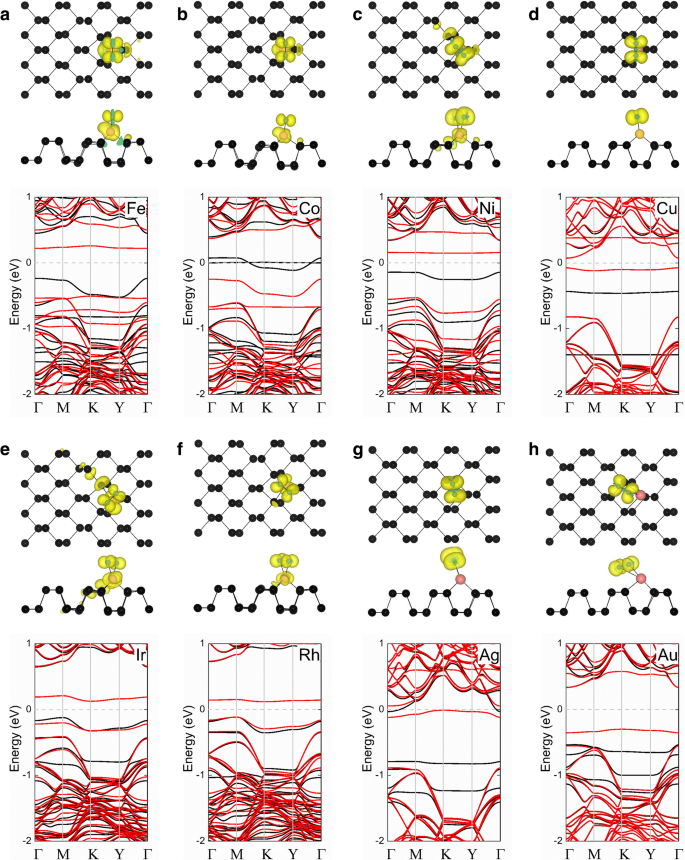
Densidades de centrifugado de a O 2 - (Fe-fosforeno), b O 2 - (Cofosforeno), c O 2 - (Ni-fosforeno), d O 2 - (Cu-fosforeno), e O 2 - (Ir-fosforeno), f O 2 - (Rh-fosforeno), g O 2 - (Ag-fosforeno) y h O 2 - Los sistemas (Au-fosforeno) se muestran en la fila superior; la estructura de bandas correspondiente de cada sistema se muestra en la fila inferior. En la fila superior, un gráfico de la densidad de carga polarizada por espín con un valor de densidad de carga iso-superficial de 0,002 e / Å 3 se superpone en las vistas superior y lateral de la estructura cristalina de fosforeno prístino; las regiones amarilla y cian corresponden a giros hacia arriba y hacia abajo, respectivamente. En los gráficos de las estructuras de bandas, las líneas negras y rojas indican canales de giro hacia arriba y hacia abajo, respectivamente; el nivel de Fermi se establece en cero y se indica mediante la línea discontinua gris
Conclusiones
Investigamos las propiedades estructurales, electrónicas y magnéticas de diferentes sistemas de TM-fosforeno. Se encontró que todos los adatoms preferían ocupar el sitio hueco en el fosforeno. La considerable energía de adsorción revela que todos los sistemas de adsorción de TM-fosforeno son bastante robustos, lo que indica que el fosforeno forma enlaces fuertes con los 12 tipos de adatomos de TM. Además, encontramos que el dopaje con Fe, Co y Au puede resultar en propiedades semiconductoras magnéticas en fosforeno monocapa, con momentos magnéticos totales de 2, 1 y 0,96 μ B , respectivamente.
Además, también examinamos las propiedades de un O 2 molécula adsorbida en el sistema TM-fosforeno. Fue muy alentador descubrir que todos los O 2 - Sistemas (TM-fosforeno), excepto para O 2 - (Pd-fosforeno), presentan una buena actividad catalítica para la oxidación del CO debido al alargamiento del enlace O – O. El O 2 - (Fe-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Ir-fosforeno) y O 2 - Los sistemas (Au-fosforeno) muestran propiedades semiconductoras de espín polarizado con momentos magnéticos de 2.00, 2.00, 1.00, 1.00, 1.14, 1.00 y 1.00 μ B . El O 2 - (Co-fosforeno) muestra características semimetálicas magnéticas, con un momento magnético de 2,00 μ B . Por tanto, nuestros resultados pueden abrir nuevas posibilidades para la aplicación de fosforeno en los campos de la catálisis y la espintrónica.
Abreviaturas
- 2D:
-
Bidimensional
- B:
-
Puente
- GGA:
-
Aproximación de gradiente generalizada
- H:
-
Sitio hueco
- LDOS:
-
Densidad local de estados
- PBE:
-
Perdew-Burke-Ernzerh
- T:
-
Parte superior de un átomo de fósforo
- TM:
-
Metal de transición
Nanomateriales
- Construcción de relés
- Electromagnetismo
- Cinta de video
- Imán
- Disquete
- Tipos de magnetómetros
- Nanodiamantes para sensores magnéticos
- Nanocluster para realizar plasmones magnéticos
- Magnetismo de percolación en nanopartículas ferroeléctricas
- Estudio de los primeros principios sobre la estabilidad y la imagen STM del borofeno
- Efectos de interacción en el ensamblaje de nanopartículas magnéticas