


Nuevos nanocompuestos de poliestireno con polianilina dopado con ácido lauril sulfúrico
Resumen
Este trabajo se concentra en la síntesis e investigación de nuevos nanocompuestos núcleo-capa de poliestireno (PS) con polianilina dopada (PANI). El látex que contiene nanopartículas de PS con tamaños de 15 a 30 nm se preparó mediante polimerización en microemulsión de estireno en medio acuoso. Los nanocompuestos de PS / PANI se sintetizaron mediante polimerización oxidativa química de anilina en el medio de látex de PS en presencia de ácido lauril sulfúrico (LSA), que sirvió como dopante y plastificante. El contenido real de PANI en los nanocompuestos sintetizados se determinó mediante el método de espectroscopía UV-Vis. La composición de los nanocompuestos y el estado de oxidación de la polianilina dopada se caracterizaron mediante espectroscopía FTIR. La morfología núcleo-capa de las nanopartículas de nanocompuestos se demostró mediante microscopía electrónica de transmisión y barrido. Se encontró que la conductividad y el comportamiento térmico en el aire de estos nanocompuestos no solo dependían de manera no lineal del contenido de polianilina dopada, sino que también se veían fuertemente afectados tanto por las propiedades plastificantes del dopante ácido como por la presencia de la cubierta de polianilina. Se ha demostrado una posibilidad de aplicación de estos nanocomposites como materiales sensores.
Antecedentes
Es bien sabido que la polianilina (PANI) tiene un conjunto único de propiedades físicas y químicas, alta estabilidad, bajo precio, etc., lo que permite sus aplicaciones multifuncionales en diferentes campos de alta tecnología como la micro y optoelectrónica, sensores y dispositivos electrocrómicos. , baterías y supercondensadores, etc. [1, 2]. La procesabilidad y aplicabilidad de PANI pueden mejorarse significativamente si se utiliza en compuestos o nanocompuestos con polímeros comunes solubles o fundibles, que pueden transformarse fácilmente en varios artículos [3]. Entre los diversos métodos de preparación de dichos materiales, la polimerización oxidativa de anilina en látex acidificados a base de agua o dispersiones que contienen nanopartículas o partículas de tamaño (sub) micrométrico de otros polímeros (estabilizadas con diferentes tensioactivos o polímeros no iónicos) se considera uno de los enfoques más eficaces [3]. Este enfoque permite obtener composites multifuncionales o nanocomposites de tipo core-shell, donde el núcleo es la (nano) partícula de polímero y la cáscara está formada por PANI [3,4,5,6,7,8,9,10]. Para facilitar la formación de la morfología núcleo-capa, la anilina se polimeriza como su sal, que aparece en el medio de látex debido a la interacción de la anilina con un dopante ácido agregado, típicamente HCl (p. Ej., [8, 10]), o como comercial sal de clorhidrato de anilina (por ejemplo, [4, 7]). En muchos casos, la estabilidad de los medios de polimerización de látex se apoya adicionalmente con aditivos estabilizadores no iónicos o iónicos (más a menudo dodecil sulfato / lauril sulfato de sodio (SDS / SLS) [3,4,5,6,7,8,9, 10]). Sin embargo, se ha desarrollado un método alternativo para preparar dichos materiales. Este método utiliza un ácido tensoactivo (p. Ej., Ácido dodecilbencenosulfónico - DBSA) que unifica las propiedades de los tensioactivos, plastificantes y dopantes ácidos y, por lo tanto, permite evitar el uso antes mencionado de HCl adicional u otro dopante ácido [11, 12].
Aspectos mecanicistas de la formación de capas o conchas de PANI en la superficie de diferentes sustratos orgánicos o inorgánicos macroscópicos (vidrio o cuarzo, películas poliméricas, fibras, etc.) y microscópicos (látex de poliestireno, sílice o titania, o partículas poliméricas, etc.) (plantillas) se han discutido en muchas publicaciones principalmente en términos de polimerización por adsorción in situ de los cationes de anilinio cargados positivamente en la superficie que típicamente tienen cargas negativas de aniones preadsorbidos / injertados / grupos funcionales [13,14,15,16,17] . Se ha aceptado generalmente que los cationes de anilinio adsorbidos polimerizan inmediatamente después de la adición del oxidante-iniciador. Naturalmente, los cationes de anilinio no adsorbidos también están involucrados en el proceso de polimerización y forman moléculas oligoméricas y poliméricas cargadas positivamente, que precipitan / adsorben en la misma superficie y, por lo tanto, provocan un aumento en el espesor de la capa de PANI.
En un enfoque alternativo, primero se añadió monómero de anilina al látex de poliestireno (PS) y se absorbió en forma neutra por las partículas del núcleo de PS durante 3 días [9]. Después de la adición del oxidante-iniciador (persulfato de amonio, APS), se forma la membrana conductora de polianilina en la interfaz de partículas y se separan ambos reactivos. Los electrones se transfieren de las moléculas de anilina a las moléculas oxidantes a través de la membrana de polianilina y, por lo tanto, la polianilina penetra gradualmente en el interior de la partícula de látex PS, en contraste con la morfología núcleo-capa antes mencionada obtenida en el recubrimiento clásico de partículas de látex con polianilina [9]. En otra variación de este enfoque, después de hinchar las partículas de PS con el monómero de anilina neutra durante 12 h, se añadieron APS y luego ácido clorhídrico al medio de reacción [18]. La adición de HCl dio como resultado la transformación de las moléculas de anilina liberadas de las partículas en cationes de anilinio, que a su vez se polimerizaron mediante oxidación química con APS. En este caso se confirmó la clara estructura núcleo-capa del compuesto formado PS / PANI [18].
El PS sirve con bastante frecuencia en materiales como el componente polimérico central debido no solo a su buena estabilidad térmica y química, características mecánicas, biocompatibilidad, etc. [19] sino probablemente a su conveniencia para la síntesis de nano / submicrones / micrones bien formados. partículas de tamaño muy adecuado para los (nano) composites con aplicaciones específicas. Por ejemplo, las partículas de PS de tamaño micrón / submicrónico recubiertas con PANI dopado se utilizaron en aceleradores electrostáticos, lo que permitió acelerar las partículas cargadas a hipervelocidades [20] o en fluidos electrorreológicos [21], etc. los compuestos [22] PS / PANI obviamente tienen potencial para aplicaciones de detección. Sin embargo, hasta donde sabemos, existe una falta de información sobre el uso de compuestos de núcleo-capa (nano) PS / PANI como materiales de detección. No obstante, recientemente se demostró que la mezcla de PS disuelto en tolueno con partículas de PANI dopadas con ácido canforsulfónico daba dispersiones adecuadas para la formación de películas compuestas sensibles al amoniaco [23]. Curiosamente, aunque estas mezclas mostraron respuestas bastante altas al amoníaco gaseoso, es decir, (Δ R / R 0 ) × 100 ~ 73% a 20 ppm, sus respuestas a concentraciones más altas de amoníaco no fueron muy diferentes y fueron sólo hasta ~ 90% a 100 ppm (Fig. 11 en [23]). Este comportamiento de concentración de respuesta débil sugiere que, debido a la preparación de la solución de estas mezclas, solo una parte de los grupos sensibles de PANI es fácilmente accesible para las moléculas de analito, y la otra parte es filtrada por la matriz de PS, lo que imparte algunas limitaciones de difusión a los materiales de detección. Por lo tanto, se puede deducir que los compuestos de núcleo-carcasa de PS / PANI con superficie PANI no apantallada pueden tener un comportamiento de detección mejorado en comparación con los materiales de mezcla preparados en solución [23].
Con base en la discusión anterior, nuestro trabajo se concentró principalmente en la síntesis de nuevos nanocompuestos núcleo-capa de nanopartículas de PS con PANI dopado con ácido lauril sulfúrico (LSA) y en la investigación de sus propiedades prácticamente importantes (morfología, estructura química, conductividad, termoestabilidad). . También se estimó su aplicabilidad potencial como materiales de detección. La elección de LSA es la característica importante de los nanocomposites, que se basa principalmente en tres requisitos previos:(1) el mismo anión tensioactivo de lauril sulfato tanto en el surfactante SLS usado en la etapa de síntesis de nanopartículas de PS central como en el dopante ácido El LSA utilizado en la etapa de síntesis de la cáscara de PANI, (2) puede actuar como el ácido protónico funcionalizado tensoactivo-dopante acidificando el medio de reacción [24, 25], y (3) forma sal de anilinio (es decir, tensoactivo reactivo monómero o surfmer) que facilitan la formación de estructuras y conchas de PANI nanométricas [25, 26].
Métodos
Materiales
La anilina (Merck) y el estireno (grado reactivo, Ucrania) se destilaron al vacío y se almacenaron bajo argón a 3-5 ° C. El persulfato de potasio oxidante (KPS) (Ucrania), el tensioactivo aniónico lauril sulfato de sodio (SLS, sinónimo de dodecilsulfato de sodio - SDS, Aldrich) eran de grado reactivo y se utilizaron sin purificación adicional. Se preparó ácido lauril sulfúrico (LSA) a partir de SLS mediante una reacción de intercambio iónico con resina KU-2-8 (Ucrania).
Preparación de látex PS
Los látex nanoparticulados de PS se prepararon mediante polimerización radical de estireno de acuerdo con el método descrito en otra parte [27]. En resumen, el estireno se polimerizó en una solución acuosa micelar de SLS con oxidante-iniciador KPS de la siguiente manera:se añadieron lentamente 2 g de estireno durante un período de 1,5 h a una solución vigorosamente agitada de 0,01 g de NaH2 PO 4 , 0,2 g SLS y 0,01 g KPS en 10 ml de agua a 70 ° C en atmósfera de argón. La mezcla se agitó durante 3 horas más a 70ºC y luego durante 1 hora más a 90ºC. La mezcla de polimerización final se enfrió a temperatura ambiente y se purificó mediante diálisis a través de membrana de celulosa con MWCO 3500 Da frente a agua destilada durante 48 h.
Preparación de nanopartículas de PS / PANI-LSA
La polimerización de anilina en el látex de PS se llevó a cabo de manera similar al método descrito en otra parte [28, 29] en las siguientes proporciones de los componentes de la mezcla de reacción:anilina / LSA =1 / 1.5 (mol / mol) y anilina / oxidante =1 / 1.25 (mol / mol) a 10 ° С. La relación en peso inicial de anilina a nanopartículas de PS en la mezcla de polimerización estaba predeterminada por las cantidades teóricas esperadas de polianilina desdopada en los nanocompuestos finales en un rango de 1 a 10% en peso. En resumen, en la primera etapa de preparación de la mezcla de polimerización, se añadió una cantidad calculada del ácido a la porción de látex de PS objetivo y se agitó a temperatura ambiente durante 30 min. En la segunda etapa, se añadió la cantidad calculada de anilina a este látex de PS acidificado seguido de agitación durante 1 h para permitir la formación completa de la sal de anilinio a temperatura ambiente y luego la mezcla preparada se enfrió a 10 ° C durante 30 min. En la tercera etapa, la cantidad calculada de la solución de KPS preenfriada a 10 ° C en agua destilada se añadió gota a gota a la mezcla de reacción seguido de agitación durante 24 ha 10 ° C. Una vez completada la polimerización de anilina, los látex de PS / PANI-LSA obtenidos se purificaron mediante diálisis a través de la membrana de celofán frente a agua destilada durante 3 días. Los nanocompuestos purificados se secaron en condiciones ambientales hasta un estado de polvo seco visualmente seguido de secado al vacío a 60ºC hasta que se alcanzó un peso constante. La muestra de PANI-LSA pura de referencia se sintetizó en las mismas condiciones en la solución acuosa en ausencia de nanopartículas de PS.
Caracterización
El contenido real de PANI en los nanocomposites sintetizados se determinó de manera similar a [29] mediante análisis de espectroscopía UV-Vis de sus soluciones en N-metil-2-pirrolidona (NMP) con la ayuda del espectrofotómetro Cary 50 (Varian). En resumen, en la primera etapa, el nanocompuesto seco se desdopó típicamente en una solución acuosa de amoniaco al 0,3% en peso durante 24 h, luego se lavó con agua destilada y luego se secó al vacío a 60 ° C hasta que se alcanzó un peso constante. En la segunda etapa, la porción fija del nanocompuesto en polvo desdopado se disolvió en NMP y se mezcló con una solución de ácido ascórbico en NMP para obtener leucoemeraldina base (LB, forma completamente reducida de PANI). En la tercera etapa, se calculó la concentración de LB a partir de la absorción UV de esta solución en una cubeta de cuarzo de 1 mm a 343 nm utilizando la curva de calibración preparada previamente. En la cuarta etapa, esta concentración de LB se volvió a calcular para el contenido real de PANI desdopado en el nanocompuesto. Este último contenido se utilizó luego para estimar el contenido de PANI-LSA dopado en este nanocompuesto de PS / PANI-LSA. Este recálculo final se basó en la relación estequiométrica teórica de LSA y nitrógenos de imina en PANI, a saber, PANI:LSA =1:0,5 de manera similar a [28, 29]. De acuerdo con [28], por simplicidad y claridad de este recálculo, postulamos el dopaje completo del PANI con solo LSA. Las composiciones de los nanocomposites sintetizados y sus notaciones se dan en la Tabla 1.
Los espectros de infrarrojos por transformada de Fourier (FTIR) de los nanocompuestos PS / PANI-LSA y las muestras de PANI-LSA puras en gránulos con KBr se registraron con una resolución de 1 cm −1 con espectrómetro Bruker Vertex 70.
Se obtuvieron imágenes de microscopía electrónica de transmisión y de barrido (TEM y SEM) con microscopios JEOL JEM-1400 e Hitachi S4800, respectivamente. Las muestras para las mediciones de TEM se prepararon colocando 2 μL de la dispersión de agua de la muestra en una rejilla de cobre de malla 200 revestida con carbón o formvar durante 15 min, seguido de una eliminación cuidadosa de la dispersión con un papel de filtro. Las muestras para las mediciones de SEM se prepararon dejando caer 5 µl de la dispersión de agua dializada de PS puro o un nanocompuesto sobre una placa de vidrio. Las muestras secas se recubrieron mediante pulverización catódica con una fina capa de oro (~ 7 nm).
La estabilidad térmica de los materiales sintetizados se estudió mediante análisis de termogravimetría (TGA) de sus muestras en el aire cuando se utilizó un sistema de derivatógrafo MOM Q-1500 D (Paulik-Paulik-Erdey) con una velocidad de calentamiento de 10 ° C / min.
Para caracterizar las propiedades de conductividad de los nanocomposites sintetizados, sus polvos se procesaron en películas tanto mediante la técnica de moldeo por compresión a 240 ° C bajo 5 MPa (usando prensa SPECAC) durante 2 min como mediante colada en placas de vidrio a partir de sus dispersiones al 3% preparadas bajo ultrasonidos.
Para estimar la aplicabilidad de los nanocompuestos PS / PANI-LSA sintetizados como materiales sensibles a gases nocivos, utilizamos el nanocompuesto NC15 más conductor y comparamos sus propiedades con PANI-LSA puro sintetizado en las mismas condiciones. Las mezclas de amoníaco-aire con concentraciones de amoníaco en el rango de 19 a 152 ppm sirvieron como analitos. Los elementos sensibles se prepararon como sigue. Un volumen de 1 μL de las dispersiones tratadas ultrasónicamente de los nanocompuestos en disolvente (2% w / v ) se fundió por gota sobre el sistema en miniatura de electrodos interdigitados de oro formados sobre el sustrato de vitrocerámica. Los elementos sensores formados se secaron a 60 ° C durante 30 min y luego se instalaron en la cámara de prueba hermética descrita en otra parte [30]. Las mezclas de aire y amoniaco preparadas se inyectaron con una jeringa en esta cámara. Las respuestas del sensor (SR) de estos elementos se registraron a temperatura ambiente y humedad relativa alrededor del 50% y se determinaron como una variación relativa de la resistencia R del sensor expuesto al analito de acuerdo con la ecuación SR =[( R - R 0 ) / R 0 ] × 100%, donde R es la resistencia de la muestra, R 0 es el valor de resistencia inicial.
Resultados y discusión
Morfología de los nanocompuestos PS / PANI-LSA sintetizados
Como se puede ver en la imagen TEM (Fig. 1a), el enfoque sintético utilizado permitió sintetizar nanopartículas de PS de forma esférica de tamaños muy pequeños en el rango de 15-30 nm. Hasta donde sabemos, estas nanopartículas de PS se encuentran entre las de PS más bajas.

TEM ( a - f ) y SEM ( g - o ) imágenes de nanocomposites PS y PS / PANI-LSA puros: a , g - PS puro; b , h - NC2; c , yo - NC3; d , m - NC6; e , n - NC11 y f , o - NC15
Las imágenes TEM de las nanopartículas de PS / PANI-LSA separadas después de la polimerización muestran que tienen tamaños que aumentan con el contenido de PANI-LSA (Fig. 1b-f). Este efecto sugiere la morfología de núcleo-capa de estas nanopartículas con núcleo de nanopartícula de PS y capa de PANI-LSA. Sin embargo, a pesar del aumento de tamaño, en el caso de contenidos bajos de PANI-LSA en los nanocomposites (NC2, NC3, NC6), es bastante difícil distinguir visualmente las conchas delgadas de PANI-LSA (Fig. 1b-d). Este problema puede explicarse probablemente por la naturaleza polimérica de ambos componentes y la estructura suelta de estas carcasas. Esto último, a su vez, puede ser causado por el gran tamaño de los aniones dopantes que dificultan la formación de conchas compactas de PANI-LSA. Sin embargo, a contenidos más altos de PANI-LSA en NC11 y especialmente en NC15, se pueden distinguir las conchas irregulares (Fig. 1e, f).
A pesar de las distribuciones de tamaño bastante amplias de las partículas nanocompuestas (Fig. 1b-f), podemos estimar aproximadamente el grosor de su capa. En particular, mientras que NC2 con el contenido más bajo de PANI-LSA (Tabla 1) contiene nanopartículas con tamaños alrededor de 15 nm similares a los del PS original, se pueden encontrar en la imagen TEM (Fig.1b) nanopartículas con tamaños de hasta 40 nm eso probablemente indica la presencia de conchas PANI-LSA con espesores de hasta 10 nm en sus superficies.
En el caso de NC3, no se observan nanopartículas de 15 nm, pero el número de nanopartículas de 30 a 40 nm con espesores de capa de hasta 10 nm aumentó significativamente (Fig. 1c). Esta tendencia se ve reforzada en las nanopartículas NC6 (Fig. 1d). Las imágenes TEM de NC11 y especialmente de NC15 muestran nanopartículas con tamaños aumentados en el rango de aproximadamente 25 a 50 nm (Fig. 1e, f). La presencia de algunas manchas de forma irregular sugiere la aparición de una fase separada de PANI-LSA en estos nanocompuestos debido a su mayor contenido. Además, la imagen NC15 permite distinguir claramente las carcasas PANI irregulares con espesores de 10 a 20 nm.
Después de limpiar y preparar el látex de PS original para la obtención de imágenes por SEM (consulte la sección "Caracterización"), las nanopartículas de PS formaron aglomerados con tamaños en el rango de 30-150 nm o más, que presumiblemente incluían 2-5 o más nanopartículas iniciales (Fig. 1g). La polimerización de anilina en el medio de látex en presencia de LSA tensioactivo cambió la situación (Fig. 1 h – o). Por lo tanto, en el contenido más bajo de PANI-LSA (1,84% en peso), se pueden ver en la imagen NC2 grandes entidades irregulares con tamaños de aproximadamente 400-500 nm que tienen una superficie bastante lisa (Fig. 1h). En el caso de NC3 con mayor contenido de PANI-LSA (3,01% en peso), las entidades tienen una tendencia a tener menos tamaños en el rango de aproximadamente 100-300 nm. Esta tendencia se ve fuertemente reforzada con un contenido más alto de PANI-LSA en NC6 (5,85% en peso). En particular, su imagen SEM muestra no solo un pequeño número de entidades con tamaños de hasta 150 nm, sino también aglomerados irregulares con tamaños en el rango de 40-100 nm (Fig. 1m). Las imágenes SEM de NC11 y NC15 (Fig. 1n, o) demuestran un mayor desarrollo de la morfología de las muestras, es decir, cambios cualitativos y cuantitativos en estos nanocompuestos debido a los contenidos más altos de PANI-LSA 11,27 y 14,82% en peso, respectivamente. Específicamente, se pueden ver aglomerados bastante densamente empaquetados con tamaños principalmente en el rango de aproximadamente 25-50 nm en la superficie plana de la muestra NC11, mientras que en el caso de la muestra NC15 aglomerados bien distinguibles de 25-50 nm dispuestos en "racimos de uvas" se observan morfología (Fig. 1n, o). Esta morfología sugiere una superficie específica más alta de NC15 en comparación con otros nanocomposites.
En general, las mediciones TEM y SEM muestran que aunque las nanopartículas de PS puro después de la limpieza tienden a aglomerarse, los bajos contenidos de PANI-LSA en los nanocompuestos NC2 y NC3 suprimen esta aglomeración. Este efecto se puede asignar a la actividad superficial de los aniones LS grandes de compensación de carga que se localizan alrededor de las capas de PANI cargadas positivamente en los núcleos de PS y, por lo tanto, separan las nanopartículas. Sin embargo, la situación se invierte en los contenidos moderados (NC6) y especialmente en los altos (NC11 y NC15) de PANI-LSA, que aparentemente facilitan la formación de conchas de PANI-LSA bastante gruesas alrededor de los núcleos de PS. Como resultado, el número de LS de compensación de carga ¯ Los aniones tanto alrededor como dentro de las carcasas PANI con carga positiva se vuelven más altos en comparación con los casos NC2 y NC3. Inevitablemente, estos aniones anfifílicos con largas colas de dodecilo pueden mejorar las interacciones intermoleculares existentes en el sistema. Estas interacciones son probablemente más fuertes que la tendencia mencionada anteriormente en NC2 y NC3 y, a su vez, pueden causar la aglomeración observada de nanopartículas NC6, NC11 y NC15.
Medidas FTIR
Las estructuras de los polímeros sintetizados se caracterizan mediante sus espectros FTIR. En particular, como se puede ver en la Fig.2, el espectro FTIR de PS contiene cinco picos característicos de vibraciones de estiramiento C – H aromáticas con el pico máximo a 3025 cm −1 [31]. Los picos de vibración de estiramiento C – H de los grupos metileno ocurren a 2920 y 2850 cm −1 . Se observan cuatro bandas de vibraciones aromáticas de estiramiento C =C a 1601, 1583, 1492 y 1452 cm −1 . Las bandas muy fuertes a 756 y 697 cm −1 puede asignarse a la vibración fuera del plano CH y la deformación fuera del plano del anillo, respectivamente, [31]. Estas bandas confirman la presencia de un grupo aromático monosustituido.
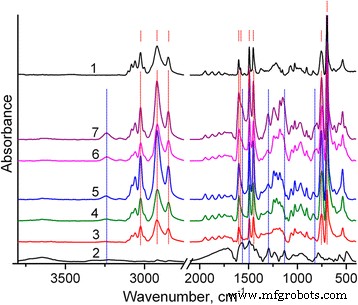
Espectros FTIR de compuestos PS (1), PANI (2) y PS / PANI-LSA:NC3 (3), NC3 (4), NC3.5 (5), NC11 (6), NC15 (7). Los picos característicos principales de PS y PANI-LSA están marcados con rayas rojas y líneas azules , respectivamente. Todas las marcas corresponden a las frecuencias discutidas en el texto
A su vez, el espectro FTIR de PANI-LSA concuerda bien con los datos publicados [32,33,34]. Contiene bandas típicas a 1565, 1492, 1294, 1133 y 818 cm −1 asignado a las vibraciones de estiramiento de los anillos de quinoides, anillos de bencenoides, estiramiento C – N en una amina aromática secundaria, modo vibratorio de un B – NH + = Q estructura, flexión C – H fuera del plano de 1.4 anillos, respectivamente. Algunas características, como un NH muy débil que estira las vibraciones en la región de 3l00–3500 cm −1 , indican que PANI se encuentra en estado dopado. Sin embargo, el B – NH + = Q intensidad de la banda a 1133 cm −1 es bastante débil, lo que sugiere un nivel de dopaje bastante bajo de este PANI-LSA [34].
Una banda distinta a aproximadamente 1180 cm −1 (Figs. 2 y 3b, curva 1) que se origina a partir de la vibración de estiramiento S =O [31] muestra que las nanopartículas de PS sintetizadas contienen aniones de lauril sulfato, que están presentes aparentemente debido a las condiciones de síntesis de las nanopartículas de PS (ver parte de Métodos). Además, se observa evidentemente un exceso de estos aniones en los compuestos finales de PS / PANI-LSA. Por tanto, como puede verse en la Fig. 3a (curva 2), las vibraciones de estiramiento C – H de los anillos aromáticos y los grupos metileno de PANI-LSA son muy débiles. Por lo tanto, las bandas intensas de C – H estirando las vibraciones de los grupos de metileno, que se revelan debido a la sustracción del espectro PS del NC15 (después de la normalización por altura de la banda a 3025 cm −1 ) (Fig. 3a, curva 4), obviamente se puede asignar a la fase SLS separada.
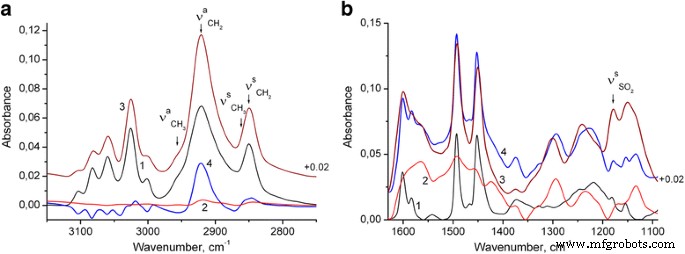
Espectros FTIR de nanopartículas de PS (1), PANI-LSA (2) y NC15 (3): a El espectro 4 es el resultado de la sustracción del espectro PS normalizado del NC15, b El espectro 4 es la suma del espectro de PS (normalizado a la altura de banda de NC15 a 3025 cm −1 ) y el espectro PANI-LSA (normalizado a la altura de banda de NC15 a 1560 cm −1 )
Para evaluar el estado de PANI en el nanocompuesto, comparamos el espectro de NC15 con el espectro del modelo (Fig. 3b, curvas 3 y 4 en consecuencia). La última es la suma de las contribuciones espectrales de PS y PANI-LSA. En particular, la contribución de PS es el espectro de PS normalizado a la altura de banda de NC15 a 3025 cm −1 (donde la absorción de PANI-LSA es muy débil), y la contribución de PANI es el espectro de PANI normalizado a la altura de banda de NC15 a 1560 cm −1 (donde la absorción de PS está ausente). Se sabe que las bandas de PANI dopadas a aproximadamente 1580 y 1490 cm −1 tienen una contribución importante de los anillos quinoide y bencenoide, respectivamente, [32,33,34]. La relación de intensidad de estas bandas es sensible a la estructura química del PANI y, por lo tanto, el predominio de los anillos quinoides sobre las unidades bencenoides en el espectro de NC15 en comparación con el espectro del modelo atestigua que el grado de oxidación de la fase PANI-LSA en el El nanocompuesto es más alto que el del PANI puro. También se puede ver que las bandas de PS del aromático C =C estirando las vibraciones a 1601 y 1583 cm −1 se amplían en el espectro NC15 y se desplazan ligeramente a longitudes de onda más bajas. Este cambio probablemente indica una interacción π – π entre PANI y PS. La intensidad de la banda NC15 a 1133 cm −1 es apreciablemente más alta que la del espectro del modelo, lo que indica la mayor conductividad de la fase PANI en este nanocompuesto en comparación con el PANI puro.
Estabilidad térmica
Recientemente, se ha demostrado para compuestos de policarbonato (PC) polímero-polímero de núcleo-capa de partículas de tamaño micrométrico con contenidos bastante bajos de PANI (~ 2% en peso de base PANI o 3.5-5.0% en peso si se dopa con ácido sulfónico aromático diferente -dopantes) que la presencia del dopante afecta fuertemente su estabilidad térmica [35]. Dependiendo del sustituyente alquilo en el anillo aromático de los dopantes, los compuestos demostraron más (sustituyente largo) o menos (sustituyente corto) disminución de su estabilidad térmica en comparación con el PC puro debido a interacciones intermoleculares específicas de los aniones dopantes grandes plastificantes y / o moléculas dopantes liberadas térmicamente con cadenas de PC [35]. Pero a una temperatura superior a 500 ° C, a la que el PANI en la cáscara se encuentra típicamente en el estado desdopado (base), la estabilidad de los compuestos fue mayor que la del PC. Este efecto se asignó a un estado específico de PANI ubicado como caparazón en la superficie del material del núcleo en los compuestos núcleo-caparazón ya una posible estabilización de la partícula del núcleo de PC por la capa de PANI [35, 36]. Con base en esta posibilidad y la morfología de los nanocompuestos, sugerimos que el efecto estabilizador de la capa de PANI también puede tener lugar para diferentes nanocompuestos núcleo-capa de polímero-PANI. La sugerencia concuerda bien con el comportamiento térmico de las nanopartículas y nanocompuestos de PS sintetizados ilustrados en la Fig. 4.
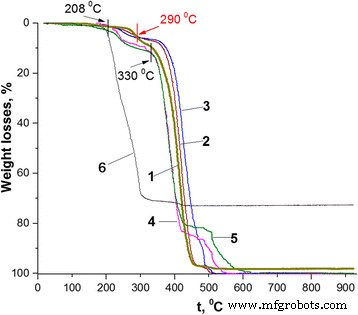
Curvas termogravimétricas de los nanocompuestos PS / PANI-LSA con diferentes contenidos de PANI-LSA (% en peso): 1 PD, 2 1,84 (NC2), 3 3.01 (NC3), 4 11.27 (NC11), 5 14,82 (NC15), 6 SLS
De hecho, las nanopartículas de PS sintetizadas demuestran una estabilidad térmica que difiere en cierta medida de la del PS a granel (compárese la curva 1 en la Fig. 4 y la Figura 1 en [37]). En particular, mientras que el último se degrada en el aire principalmente en un solo paso de 200 a 450 ° C [37], la curva termogravimétrica (TG) del primero muestra aproximadamente tres etapas:pérdidas de peso semanales (~ 1,9% en peso) desde el principio hasta el 262 ° C, el segundo en el rango de 262–330 ° C y el tercero en el rango de 330–505 ° C. Esta diferencia probablemente se puede explicar por la especificidad de la síntesis de nanopartículas de PS que dio como resultado la presencia inevitable en su composición de impureza de SLS que a su vez cambió el comportamiento térmico de PS. Esta sugerencia concuerda bien con el hecho de que la temperatura de degradación final de SLS está muy cerca del comienzo (330 ° C) de la tercera etapa (principal) de la degradación de las nanopartículas de PS (Fig. 4, curvas 1 y 6).
Como se puede ver en la Fig. 4, las curvas TG de los nanocomposites tienen una forma similar a la del PS y, además, demuestran pequeñas pérdidas de masa similares a temperaturas de hasta 120 ° C, que normalmente se pueden asignar a la evaporación del agua [35]. . A temperaturas más altas, se pueden ver diferencias significativas en la estabilidad térmica de las muestras con cargas bajas y altas de PANI-LSA, que, en general, brindan información complementaria sobre la especificidad del comportamiento térmico de los compuestos conocidos que contienen PANI núcleo-capa. En particular, tres de los nanocompuestos (NC2, NC3 y NC11) con contenido de PANI-LSA ≤ 11,27% en peso muestran una estabilidad térmica similar a la del PS hasta 208 ° C (Fig. 4, Tabla 2). Sin embargo, NC15 con el contenido más alto de PANI-LSA (14,82% en peso) es menos estable que PS incluso a 120 ° C (Fig.4, curvas 1 y 5; Tabla 2) que probablemente también se puede asignar a la evaporación no solo de humedad. pero también probablemente al dopante no unido y / o las impurezas de monómero / oligómero que no reaccionan [38].
En el rango de temperatura de 208-262 ° C, todos los nanocomposites muestran pérdidas de peso, que son mayores que los contenidos de LSA pero significativamente menores que las pérdidas de peso de PS (Fig. 4, curvas 1, 4 y 5, Tabla 2). Sin embargo, en el caso de NC2 y NC3, estas pérdidas son incluso mayores que los contenidos de PANI-LSA. Con base en la alta estabilidad térmica de la base PANI [39] y el comportamiento térmico de las nanopartículas de PS (Fig. 4, curva 1), probablemente podamos asignar las pérdidas de nanocompuestos no solo a la evaporación y degradación del dopante sino también a la degradación del Componente PS. Además, mientras que las pérdidas de peso de NC2 y NC3 a 262 ° C (Tabla 2) exceden las sumas de su contenido de LSA y la pérdida de PS (3.02 y 3.7, respectivamente), se puede suponer que alguna mejora de la degradación termooxidativa del componente central de PS de los nanocomposites pueden ser causados por productos de degradación del dopante.
Aunque las pérdidas de nanocomposites normalmente aumentan a temperaturas más altas, a 290 ° C las curvas TG de NC2 y NC3 (a diferencia de las de NC11 y NC15) se cruzan con la curva TG de PS en el punto de 5,58% en peso (Fig.4, Tabla 2). . Este comportamiento, en general, sugiere una pérdida completa del dopante [35, 37, 38] y la transformación del componente PANI-LSA en el PANI desdopado. Above this, temperature NC2 and NC3 are more stable than PS up to the end of the heating process (Fig. 4, curves 1–3). As a consequence, the position of the TG trace of PS nanoparticles along the temperature axis in the range of 262–430 °C roughly separates positions of the nanocomposites with low and high contents of PANI-LSA (Fig. 4). This fact confirms a difference which is probably inherent to these two sets of nanocomposites.
Indeed, one can see strongly different course of the thermal degradation of these nanocomposites both in the range of 262–430 °C and above 430 °C. Whereas all these nanocomposites have the core-shell morphology, it is unlikely that only this morphological factor can explain their specific thermal behavior. However, if to take into account the presence in their composition of the LSA dopant, which contains the long dodecyl tail with plasticizing ability [3], we can at least partially understand such difference as a result of intermolecular interactions (causing a plasticizing effect [40]) of the dopant anion with the polymer components of the nanocomposites. Naturally, in the case of low or high contents of the PANI-LSA component, its influence on thermal behavior of the nanocomposites will be less (NC2, NC3) or more (NC11, NC15) significant. In the latter case, the plasticization effect is so strong that the thermogramms of NC11 and NC15 (Fig. 4, TG curves 4 and 5) take positions below the PS one up to 430 °C even after a complete removal of the dopant (above ~ 290 °C) because of weakened interactions between PS macromolecules. Slowing down the degradation rate of the nanocomposites with high content of PANI base at temperatures above 430 °C can be probably explained by cross-linking of its chains [39] and possible enhancement of the stabilizing role of the PANI shell.
In the case of NC2 and NC3, the situation is obviously opposite to NC11 and NC15. In particular, contents of PANI-LSA are quite small, and therefore, quantities of the plasticizing dopant LSA are not enough to significantly weaken interactions between PS macromolecules. As a consequence, once the dopant is eliminated completely, the nanocomposites display thermostability which is higher than that of PS nanoparticles (Fig. 4, curves 2 and 3, intersection point at 290 °C). In spite of the low content of PANI-LSA and, therefore, of its thin shell, these NC2 and NC3 behaviors match well with the suggestion about stabilizing effect of the PANI base shell.
Conductivity and sensing properties of the synthesized nanocomposites
One of most important features of polymer-polymer composites, in particular of PANI-containing composites, is probably their ability to withstand conditions of common treatments, which are typically applied to produce different articles. Therefore, a lot of studies have been performed to estimate changes in properties of these materials after treatments by melting or solution techniques [3, 38, 39]. Based on these studies and the thermally induced weight losses of the synthesized nanocomposites (Table 2, Fig. 4), one might expect that such important property of doped PANI as conductivity could be changed under these treatments. Indeed, as one can see from Fig. 5, the values of conductivity of the cast and compression molded PS/PANI-LSA films strongly differ. To quantify the difference, we treated these data (Fig. 5) by the scaling law based on the percolation theory [41] in accordance with the known methodology of processing the conductivity behavior of polyaniline networks in composites [42, 43]:
$$ \sigma ={\sigma}_{\mathrm{o}}{\left(f-{f}_c\right)}^t $$ (1)donde σ o is the constant displaying conductivity of the PANI conducting phase, f is the volume fraction of PANI, f c is the percolation threshold, and t is the critical exponent. Volume fractions of PANI-LSA in the nanocomposites were calculated on the basis of densities of PS and PANI-LSA, i.e., 1.04 [44] and 1.18 g/cm 3 [45], respectively.

Dependencies of DC conductivity of the cast (1 ) and compression molded (2 ) PS/PANI-LSA nanocomposite films on the volume fraction of PANI-LSA
The power-law dependence was determined with various trial values of f c by applying a linear regression analysis to the plot of log σ versus log (f − f c ). The solid lines represent best fits to the data with the correlation coefficients of 0.996 and 0.993 for the cast and compression molded nanocomposite films, respectively.
The observed nonlinear dependences (Fig. 5) are obviously the result of formation of the phase-separated conducting percolation network of PANI-LSA in the bulk of the nanocomposite films. It is interesting to note that the percolation thresholds are quite low (f c = 1.26%) and independent on the used processing techniques. This f c value is significantly lower than the theoretical model suggests for a random lattice of spheres (from 15 to 30% depending on the sphere diameter) [41]. However, the conductivity of the PANI conducting phase (σ o ) in the cast nanocomposite films is more than two and a half times higher than that of the compression molded ones (2.3 × 10 −4 and 8.9 × 10 −5 S/cm, respectively). Obviously, the lower conductivity of the conducting phase in the compression molded film is caused by the partial thermal degradation of PANI-LSA under the melting treatment temperature (240 °C). The values of the critical exponent t for the cast and compression molded films are 1.14 and 2.62 accordingly. Such inequality in the critical exponent indicates a strong difference in the spatial structure of the percolation cluster, which results in the different slopes of the curves. As a consequence, the conductivities of the cast nanocomposite films are more than three orders of magnitude higher than those of the compression molded ones at low volume fractions (contents) of PANI-LSA (Fig. 5).
Nevertheless, despite the significant difference of the cast and compression molded films, one can deduce that the conductivity level is enough to apply the both materials for antistatic applications. On the other hand, the obtained conductivity values of the synthesized nanocomposites are significantly lower (by 2–3 orders of magnitude) than in the case of the similar core-shell submicron/micron-sized PS/PANI composites [4, 7, 9, 14]. To understand this difference and to improve the conductivity of these new PS/PANI-LSA nanocomposites, new studies are planned. However, one can suggest that non-optimal conditions of preparation of these new materials are at least partial explanation of this low conductivity level.
Based on better conductivity properties of the cast PS/PANI-LSA films and known high sensing ability of doped PANI [46], we estimated their potential as sensing materials to determine concentrations of ammonia in its gaseous mixtures with air. The measurements were performed on the example of the films of NC15 and pure PANI-LSA cast on electrodes of the transducer (see “Methods” chapter).
Both films demonstrate quite high sensitivity to ammonia in the range of 19–152 ppm (Fig. 6). However, while NC15 is more sensitive to ammonia than pure PANI-LSA in the concentration range of 19–114 ppm, at higher concentrations, the situation becomes opposite.

Sensor responses (calibration curves) of the cast pure PANI-LSA (1 ) and NC15 (2 ) films to different concentrations of ammonia in the mixtures with air
The better efficiency of NC15 in this narrowed ammonia concentration range can be probably assigned to core-shell morphology of the nanoparticles constituting the cast nanocomposite film. This morphology typically specifies higher surface of the film as compared with pure PANI-LSA and improves sensitivity of sensing materials [25, 28]. The enhancement of the sensing responses of pure PANI-LSA at ammonia concentrations above 114 ppm (Fig. 6, curve 2) can be probably assigned to additional involving in the sensing process of the PANI-LSA clusters located under the surface of the film. Naturally, the quantity of these clusters in the pure doped PANI film is much higher than in the case of the thin PANI-LSA shells on the core particles constituting the nanocomposite film. Therefore, their involvement in the sensing process inevitably increases sensor responses of the pure doped PANI film as compared with the NC15 one.
Conclusions
The new PS/PANI-LSA nanocomposites have been synthesized with the core-shell nanoparticle sizes ~ 25–50 nm, which to our knowledge are the lowest ones among the similar composites published elsewhere. The use of LSA as acidifying agent for the aniline containing PS latex medium and addition of the oxidant resulted in the precipitation of the thin PANI-LSA shell (~ 10–20 nm) on the surface of the PS nanoparticles (synthesized in the presence of SLS). As a consequence, both the shell and PS core contained the same lauryl sulfate surface active anion unlike the known core-shell PS/PANI composites synthesized with a PS latex surfactant-stabilizer and PANI dopant of different nature.
We have found that although the synthesized very small PS nanoparticles (15–30 nm) after cleaning tend to agglomerate in the dry state, the low contents of PANI-LSA in the nanocomposites suppress this agglomeration probably due to the surface activity of charge compensating large LS¯ anions which localize around positively charged PANI shells on PS cores and separate the nanoparticles. However, the situation becomes opposite at the moderate and especially at the high PANI-LSA contents, which apparently facilitate formation of quite thick PANI-LSA shells around PS cores. In this case, a number of charge compensating LS ¯ anions both around and inside of the positively charged PANI shells is higher as compared with the low contents of PANI-LSA in the nanocomposites. As a consequence, these amphiphylic anions with long dodecyl tails can enhance intermolecular interactions in the system and lead to the agglomeration of the nanoparticles with high contents of PANI-LSA.
A possibility of such agglomeration effects should be taken into account when using similar nanocomposites in applications which need charged nanoparticles [7, 20, 25]. We believe that applied in this work method of determination of the real PANI content in the PS/PANI nanocomposites can allow better control of their properties.
Based on FTIR and conductivity studies of the synthesized nanocomposites, we proved that oxidation state and conductivity of the PANI phase are appreciably higher than those of pure PANI-LSA. Moreover, we demonstrate here that thermal behavior of these nanocomposites in air is strongly different for low and high PANI-LSA loadings that probably stems both from the plasticizing ability of the LSA dopant and stabilizing effect of the PANI shell. This fact, in general, gives the complementary information about the thermal behavior specificity of the known PANI containing core-shell composites.
At the same time, based on thermal stability, conductivity and sensor studies, we conclude that properties of the synthesized PS/PANI-LSA nanocomposites testify to their potential applicability as materials for antistatic and sensing applications.
Nanomateriales
- ACEO® presenta una nueva tecnología para la impresión 3D con silicona
- Llevando la industria aeroespacial a nuevas alturas con la impresión 3D (2020)
- Los módulos de computadora se vuelven miniatura con el nuevo estándar OSM
- congatec:nuevo módulo SMARC con mini procesador NXP i.MX 8M
- Creación rápida de prototipos de SLA con la nueva resina de borrador
- Algo nuevo:Cápsulas de café en PLA con IML
- Honeywell impulsa el IIoT de energía con una nueva asociación
- Omron presenta un nuevo robot industrial con IA integrada
- 5G con IoT:una nueva era en la digitalización
- Exploración de nichos con nuevas tecnologías EDM
- Seleccionar etiquetas de activos es más fácil con nuevos recursos